














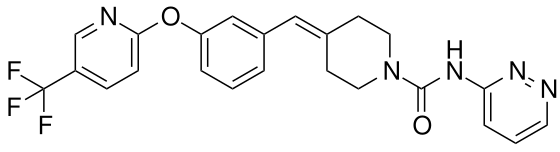
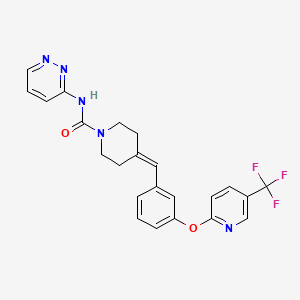
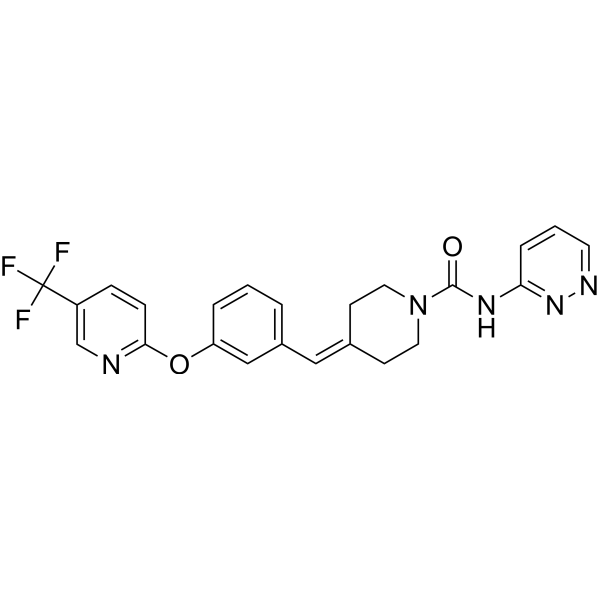
Redafamdastat
cas 1020315-31-4, PF 04457845
JZP-150; JZP150; PF-04457845; PF-4457845; PF04457845; PF4457845, Q7119045
WeightAverage: 455.441
Monoisotopic: 455.156909393
Chemical FormulaC23H20F3N5O2
- 1-Piperidinecarboxamide, N-3-pyridazinyl-4-[[3-[[5-(trifluoromethyl)-2-pyridinyl]oxy]phenyl]methylene]-
- N-pyridazin-3-yl-4-[[3-[5-(trifluoromethyl)pyridin-2-yl]oxyphenyl]methylidene]piperidine-1-carboxamide
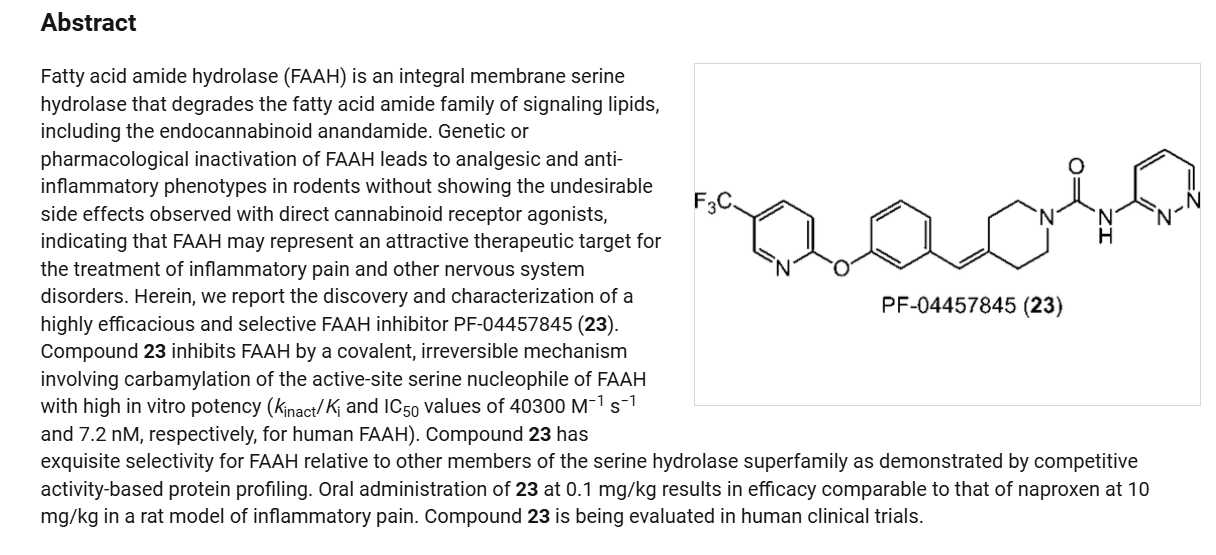
Redafamdastat (INNTooltip International Nonproprietary Name; developmental code names JZP-150, PF-04457845) is an inhibitor of the enzyme fatty acid amide hydrolase (FAAH), with an IC50Tooltip half-maximal inhibitory concentration of 7.2 nM, and both analgesic and anti-inflammatory effects in animal studies comparable to those of the cyclooxygenase inhibitor naproxen.[1] It was being developed by Jazz Pharmaceuticals for the treatment of alcoholism, pain, and post-traumatic stress disorder (PTSD) and reached phase 2 clinical trials.[2][3] However, development of the drug was discontinued in December 2023.[2]
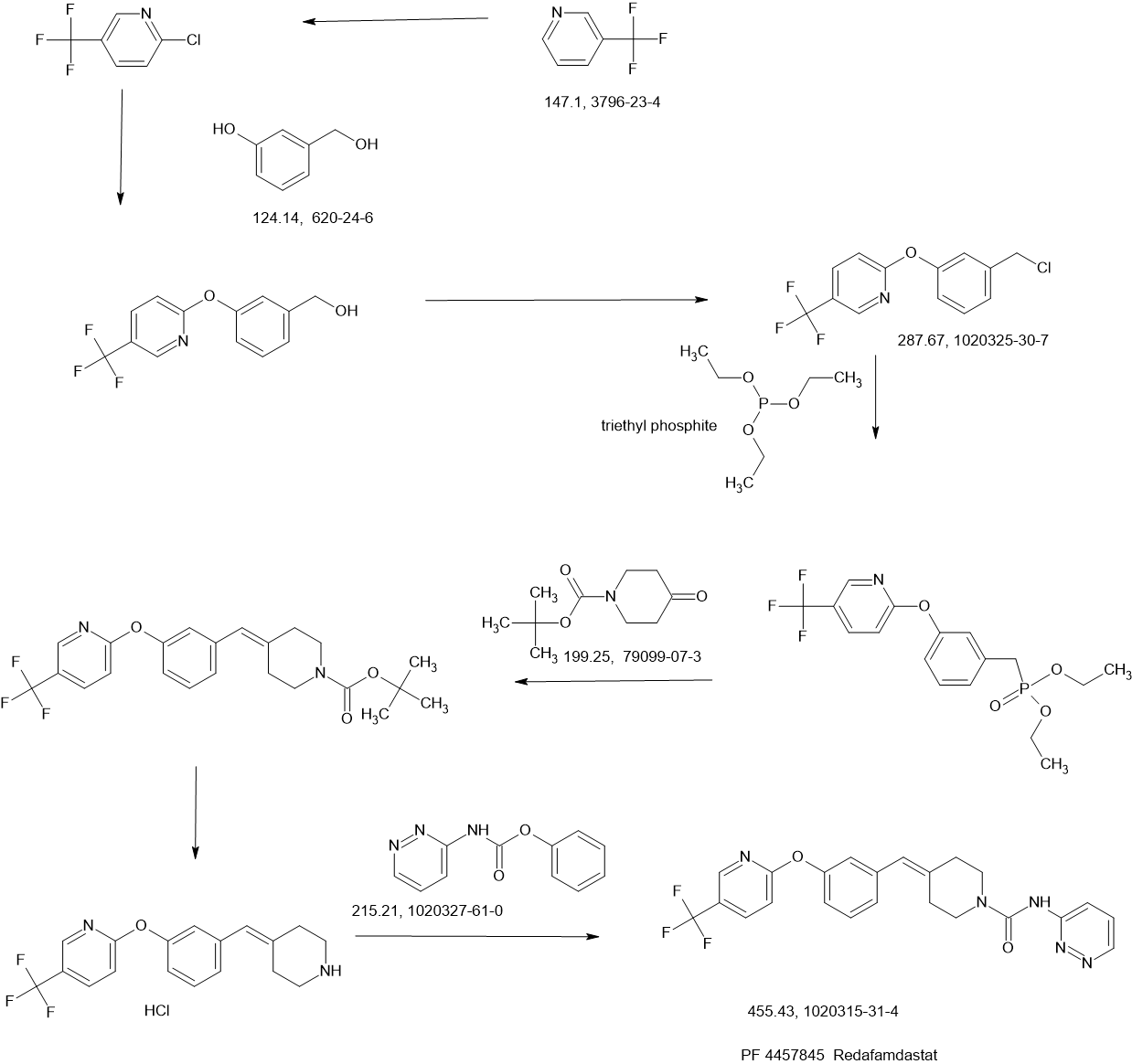
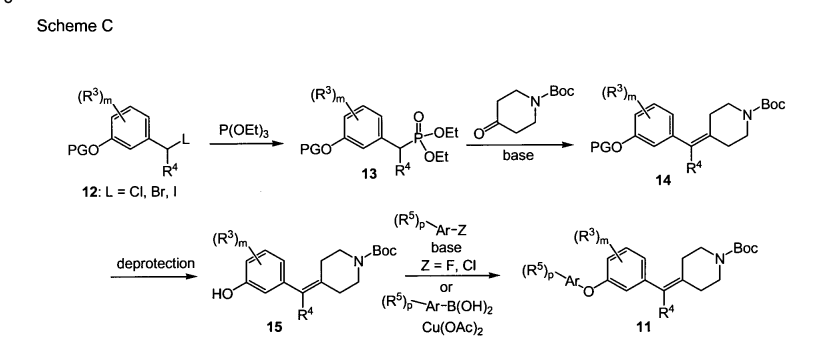
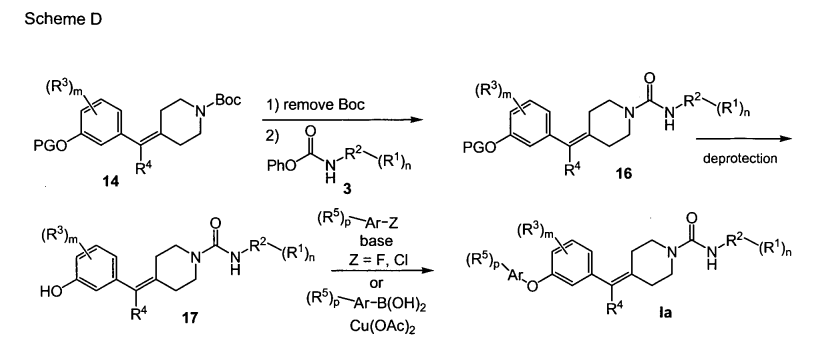
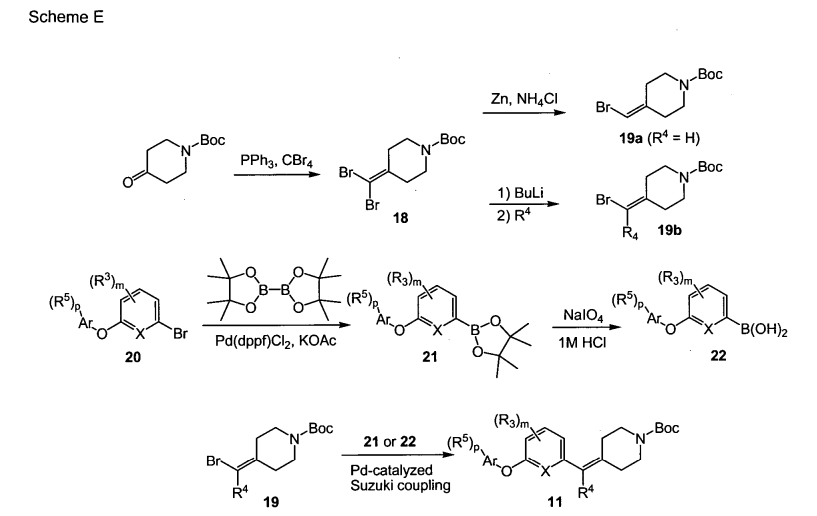
SCHEME
PAPER
ACS Medicinal Chemistry Letters (2011), 2(2), 91-96 86%
PATENT
WO2008047229 86%
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2008047229&_cid=P11-MCY7EC-33944-1
Example 1a
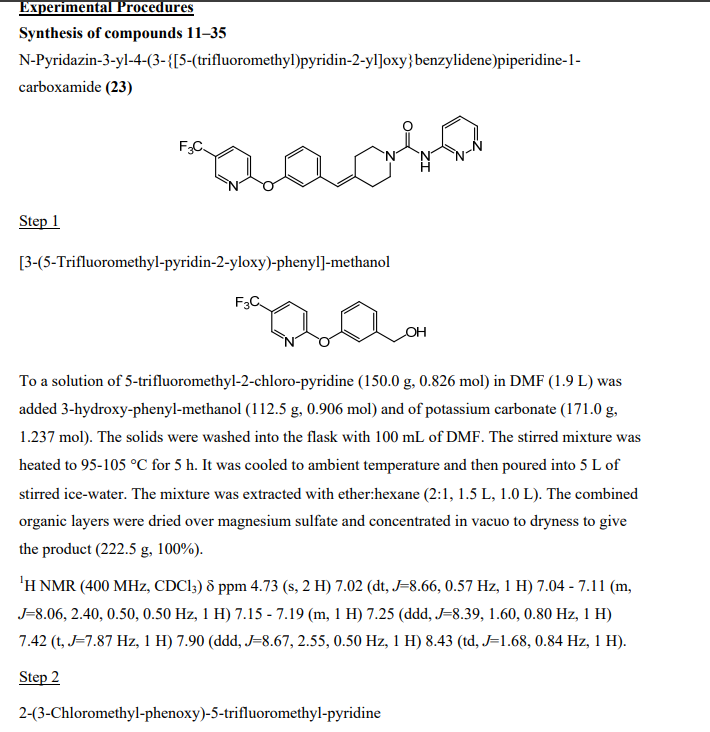
Synthesis of N-pyridin-3-yl-4-(3-(f5-(trifluoromethyl)pyridin-2-ylloxy)benzylidene)piperidine-1-carboxamide
Phenyl pyridin-3-ylcarbamate
To a stirred solution of 3-aminopyridine (51.7 g, 0.549 moles) in THF (900 mL) at -10 0C was added pyridine (52.1 g, 0.659 moles) in a stream over a 10 min period, followed by the dropwise addition of phenyl chloroformate (90 g, 0.575 moles) over a 20 min period. The reaction tempature increased to 5 0C. A precipitate formed during the addition. The resulting suspension was stirred at temperatures reaching ambient temperature over the next 3 h. The reaction mixture was partitioned between water (2 L) and EtOAc (1.5 L). The aqueous portion was extracted with EtOAc (1 L). The combined organic portions were dried (MgSO4) and concentrated in vacuo to a damp solid residue. This was suspended in
EtOAc:ether (1 :1 , 600 ml_). The resulting suspension was stirred at -10 0C for 2 h and filtered. The solid was rinsed with EtOAσether (1 :1 , 100 ml.) and pressed dry under suction. Further drying in vacuo at 35 0C for 7 h provided 104 g (88%) of product. Analysis, Calcd for Ci2H10N2O2: C, 67.28; H, 4.71 ; N, 13.08. Found: C, 67.15; H, 4.76; N, 12.87.
Step i
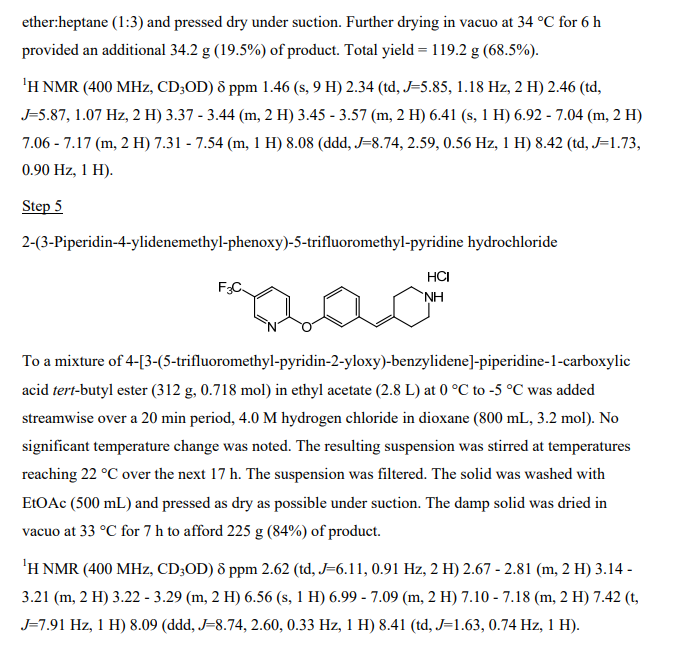
[3-(5-Trifluoromethyl-pyridin-2-yloxy)-phenyl]-methanol
3-Hydroxymethyl-phenol (5.00 g, 40.3 mmol, from Lancaster Synthesis), 2-chloro-5-trifluoromethyl-pyridine (7.31 g, 40.3 mmol, from TCI America) and potassium carbonate (6.96 g, 50.3 mmol) were suspended in dimethylformamide (80 mL) and heated to 95 0C. After stirring for 16 h, the solvent was distilled off in vacuo at 65 0C, and a residue was partitioned between water and heptane/ethyl acetate (1 :1 ). The organic layer was separated and the aqueous was extracted again with heptane/ethyl acetate (1 :1 ). The combined organic layer was dried over sodium sulfate, filtered and concentrated to give a residue. The residue was purified by silica gel chromatography (10-60%, EtOAc:heptane) to afford the desired product (5.70 g, 53% yield) as a light yellow oil. 1H NMR (400 MHz, CDCI3) δ ppm 4.73 (s, 2 H) 7.02 (dt, J=8.66, 0.57 Hz, 1 H) 7.04 – 7.11 (m, J=8.06, 2.40, 0.50, 0.50 Hz, 1 H) 7.15 – 7.19 (m, 1 H) 7.25 (ddd, J=8.39, 1.60, 0.80 Hz, 1 H) 7.42 (t, J=7.87 Hz, 1 H) 7.90 (ddd, J=8.67, 2.55, 0.50 Hz, 1 H) 8.43 (td, J=1.68, 0.84 Hz, 1 H).
Step 2
2-(3-Chloromethyl-phenoxy)-5-trifluoromethyl-pyridine
[3-(5-Trifluoromethyl-pyridin-2-yloxy)-phenyl]-methanol from Step 1 (4.68 g, 17.4 mmol), in
dichloromethane (46 mL), was cooled to 0 0C, and treated dropwise with thionyl chloride (1.40 mL, 19.1 mmol). The reaction mixture was allowed to warm to ambient temperature and was stirred for 30 rηjn. Toluene (10 mL) was added and the mixture was concentrated by evaporation to form a residue. The residue was evaporated again from toluene and dried under high vacuum to afford the desired product (4.88 g, 98% yield) as an oil. 1H NMR (400 MHz, CDCI3) δ ppm 4.60 (s, 2 H) 7.03 (d, J=8.70 Hz, 1 H) 7.11 (ddd, J=8.09, 2.35, 0.94 Hz, 1 H) 7.20 (t, J=2.03 Hz, 1 H) 7.26 – 7.31 (m, 1 H) 7.42 (t, J=7.88 Hz, 1 H) 7.91 (dd, J=8.67, 2.53 Hz, 1 H) 8.44 (dd, J=1.51 , 0.90 Hz, 1 H).
Step 3
[3-(5-Trifluoromethyl-pyridin-2-yloxy)-benzyl]-phosphonic acid diethyl ester
2-(3-Chloromethyl-phenoxy)-5-trifluoromethyl-pyridine (4.88 g, 17.0 mmol) from Step 2 was treated neat with triethylphosphite (4.36 mL, 25.4 mmol) and heated to 150 0C. After 6 h, the reaction mixture was cooled, treated with an additional 0.5 mL triethylphosphite (2.9 mmol) and reheated to 150 0C. After 6 h, the reaction mixture was removed from the heat and slowly treated with heptane (about 60 mL) while stirring to afford a white solid. The solid was collected by filtration, washed with heptane and dried in a vacuum oven for 16 h at 45 0C to afford a white powder (5.99 g, 91% yield). MS (APCI) M+1= 390.1 ; 1H NMR (400 MHz, CDCI3) δ ppm 1.26 (t, J=7.02 Hz, 6 H) 3.18 (d, J=21.83 Hz, 2 H) 3.99 – 4.10 (m, 4 H) 7.01 (d, J=8.58 Hz, 1 H) 7.03 – 7.08 (m, 1 H) 7.12 (q, J=2.21 Hz, 1 H) 7.19 – 7.24 (m, 1 H) 7.38 (t, J=7.90 Hz, 1 H) 7.90 (dd, J=8.58, 2.53 Hz, 1 H) 8.43 (dd, J=1.66, 0.88 Hz, 1 H).
Step 4
4-[3-(5-Trifluoromethyl-pyridin-2-yloxy)-benzylidene]-piperidine-1 -carboxylic acid tert-butyl ester
[3-(5-Trifluoromethyl-pyridin-2-yloxy)-benzyl]-phosphonic acid diethyl ester (2.3 g, 6.0 mmol) from Step 3 and 1 ,4,7,10, IS-pentaoxacyclopentadecane (15-Crown-5, 0.03 ml_, 0.15 mmol) were combined in THF (10 ml_). The mixture was cooled to 0 °C and sodium hydride (240 mg, 60% dispersion in mineral oil, 6.0 mmol) was added. The reaction was warmed to room temperature, stirred for 30 minutes and then cooled back to 0 0C. A solution of 4-oxo-piperidine-1-carboxylic acid tert-butyl ester (1.2 g, 6.0 mmol) in THF (6 ml_) was added and the reaction was warmed to room temperature. After 16 hours, water was added and the layers were separated. The aqueous layer was extracted with EtOAc (2X200 mL) and the combined organic layer was dried over anhydrous sodium sulfate, filtered and concentrated to a thick oil. Treatment of the oil with hot isopropyl ether (45 mL) provided the title compound as a white solid (1.88 g). 1H NMR (400 MHz, CD3OD) δ ppm 1.46 (s, 9 H) 2.34 (td, J=5.85, 1.18 Hz, 2 H) 2.46 (td, J=5.87, 1.07 Hz, 2 H) 3.37 – 3.44 (m, 2 H) 3.45 – 3.57 (m, 2 H) 6.41 (s, 1 H) 6.92 – 7.04 (m, 2 H) 7.06 – 7.17 (m, 2 H) 7.31 – 7.54 (m, 1 H) 8.08 (ddd, J=8.74, 2.59, 0.56 Hz, 1 H) 8.42 (td, J=1.73, 0.90 Hz, 1 H).
Step 5
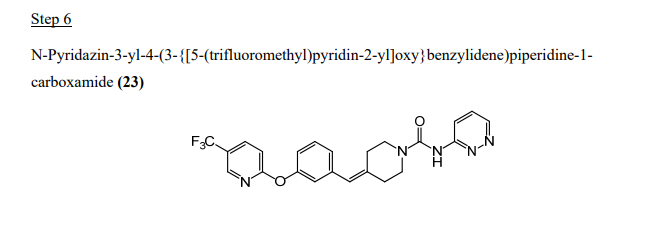
2-(3-Piperidin-4-ylidenemethyl-phenoxy)-5-trifluoromethyl-pyridine hydrochloride
4-[3-(5-Trifluoromethyl-pyridin-2-yloxy)-benzylidene]-piperidine-1-carboxylic acid tert-butyl ester (1.35 g, 3.11 mmol) from Step 4 was dissolved in CH2CI2 (30 mL) and treated with HCI in diethyl ether (10 mL, 2.0 M, 20 mmol). After 16 hours the reaction was concentrated in vacuo to form a residue and the residue was suspended in acetonitrile (10 mL) to yield a solid. Filtration of the solid provided the title compound as a white solid (1.1 g). 1H NMR (400 MHz, CD3OD) δ ppm 2.62 (td, J=6.11 , 0.91 Hz, 2 H) 2.67 – 2.81 (m, 2 H) 3.14 – 3.21 (m, 2 H) 3.22 – 3.29 (m, 2 H) 6.56 (s, 1 H) 6.99 – 7.09 (m, 2 H) 7.10 – 7.18 (m, 2 H) 7.42 (t, J=7.91 Hz, 1 H) 8.09 (ddd, J=8.74, 2.60, 0.33 Hz, 1 H) 8.41 (td, J=1.63, 0.74 Hz, 1 H).
Step 6
2-(3-Piperidin-4-ylidenemethyl-phenoxy)-5-trifluoromethyl-pyridine hydrochloride (800 mg, 2.16 mmol, from Step 5), phenyl pyridin-3-ylcarbamate (508 mg, 2.37 mmol) and diisopropylethylamine (0.75 mL, 4.52 mmol) were combined in acetonitrile (10 mL) and stirred at room temperature. After 16 hours, the reaction was concentrated forming a residue and the residue was partitioned between EtOAc and water. The organic layer was separated, washed with 5% NaOH (aq), dried over anhydrous sodium sulfate, filtered and concentrated. Treatment of the residue with hot isopropyl ether and purified from isopropyl ether/methanol provided the title compound as a white solid (574 mg). MS (APCI 10V) AP+ 455.3, 376.2, 335.2, AP- 453.2; 1H NMR (400 MHz, CD3OD) δ ppm 2.46 (td, J=5.86, 0.97 Hz, 2 H) 2.58 (td, J=5.82, 1.16 Hz, 2 H) 3.51 – 3.60 (m, 2 H) 3.61 – 3.70 (m, 2 H) 6.46 (s, 1 H) 6.98 – 7.07 (m, 2 H) 7.09 – 7.19 (m, 2 H) 7.34 (ddd, J=8.41 , 4.81 , 0.65 Hz, 1 H) 7.40 (td, J=7.69, 0.74 Hz, 1 H) 7.91 (ddd, J=8.38, 2.58, 1.44 Hz, 1 H) 8.08 (ddd, J=8.73, 2.61 , 0.55 Hz, 1 H) 8.16 (dd, J=4.84, 1.06 Hz, 1 H) 8.43 (td, J=1.74, 0.91 Hz, 1 H) 8.58 (d, J=1.88 Hz, 1 H).
Example 1b
Large scale synthesis of N-pyridin-3-yl-4-(3-{[5-(trifluoromethyl)pyridin-2-ylloxy)benzylidene)piperidine-1- carboxamide
Step 1 : Preparation of r3-(5-Trifluoromethyl-pyridin-2-yloxy)-phenyll-methanol
To a solution of S-trifluoromethyl^-chloro-pyridine (150.0 g, 0.826 mol) in DMF (1.9 L) was added 3-hydroxy-phenyl-methanol (112.5 g, 0.906 mol) and of potassium carbonate (171.0 g, 1.237 mol). The solids were washed into the flask with 100 mL of DMF. The stirred mixture was heated to 95-105 0C for 5 h. It was cooled to ambient temperature and then poured into 5 L of stirred ice-water. The mixture was extracted with etheπhexane (2:1 , 1.5 L, 1.0 L). The combined organic layers were dried over magnesium sulfate and concentrated in vacuo to dryness to give the product (222.5 g, 100%).
Step 2: Preparation of 2-(3-chloromethyl-phenoxy)-5-trifluoromethylpyridine
To a solution of [3-(5-trifluoromethyl-pyridin-2-yloxy)-phenyl]-methanol (281.0 g, 1.044 moles) in dichloromethane (2.0 L) at -5 0C was added dropwise over a 25 min period thionyl chloride (136.6 g, 1.148 mol). A few minutes into the addition, a white substance separated but this went into solution several minutes later. The reaction was stirred at ambient temperature for 1 h and then was concentrated in vacuo to near dryness (357 g). 200 mL of toluene was added to the residue and the solution was again concentrated in vacuo to near dryness. 200 mL of toluene was added and some solid (-8 g) was filtered off. The filtrate was concentrated in vacuo to -390 g of dark yellow liquid.
Step 3: Preparation of [3-(5-trifluoromethyl-pyridin-2-yloxy)-benzyll-phosphonic acid diethyl ester
A solution of 2-(3-chloromethyl-phenoxy)-5-trifluoromethylpyridine (-298 g, -1.036 mol) containing some toluene in triethyl phosphite (267.0 g, 1.551 mol) was heated to 135 °C-140 0C for 7 h. Boiling began at -110 0C and continued throughout the reaction. The solution was left standing at ambient temperature overnight and it solidified. The solid was suspended in etheπhexane (1 :2, 450 mL), and the suspension was stirred at ambient temperature for 3 h and filtered. The solid was rinsed with etherhexane (1 :2, 150 mL) and pressed dry under suction. Further drying in vacuo at 32 0C for 7 h provided 286.3 g (71 % – 2 steps from crude chloride) of product. The filtrate was concentrated in vacuo to remove the low boiling solvents. Triethyl phosphite (36.0 g, 0.217 mol) was added and the solution was heated to 130 0C for 2 h. The reaction was cooled to 100 0C and 300 mL of heptane was added slowly. A solid separated. As the temperature decreased to -30 0C, 150 mL of ether was added. The resulting suspension was left standing at ambient temperature overnight and was filtered. The solid was rinsed with etherheptane (1 :2, 75 mL) and pressed dry under suction. Further drying in vacuo at 32 0C for 7 h afforded and additional 35.7 g (9%) of product. Total yield = 322 g (80%). Anal. Calcd for C17H19 F3NO4P (389.31 ) : C, 52.45; H, 4.92; N, 3.60; F, 14.64; P, 7.96. Found : C, 52.73; H, 5.04; N, 3.58; F, 14.35; P, 7.74; chloride, <0.10%.
Step 4: Preparation of 4-f3-(5-trifluoromethyl-pyridin-2-yloxy)-benzylidenel-piperidine-1-carboxylic acid tert-butyl ester
To a stirred mixture of [3-(5-trifluoromethyl-pyridin-2-yloxy)-benzyl]-phosphonic acid diethyl ester (155.7 g, 0.40 mol) in tetrahydrofuran (800 mL) at -10 0C was added dropwise over a 5 min period 1.0 M tBuOK in tetrahydrofuran (420.0 mL, 0.42 mol). The temperature rose to -3 °C during the addition. The resulting red mixture was stirred between -6 0C and -10 0C for 2.5 h. A solution of tert-butyl 4-oxopiperidine-1-carboxylate (79.7 g, 0.40 mol) in tetrahydrofuran (300 mL) was added dropwise over a 5 min period. The temperature rose to 2 0C. The resulting red mixture was stirred at temperatures reaching 21 0C over the next 16 h. TLC showed product with no phosphonate present. The mixture was poured into 3.5 L of stirred ice-water. The resulting suspension was stirred at ambient temperature for 2.5 h and then was extracted with successive 1.0 L and 0.6 L portions of dichloromethane. The combined extracts were washed with 500 mL of brine, dried over magnesium sulfate and concentrated in vacuo to a thick semi solid residue. 250 mL of methyl t-butyl ether was added. The suspension was stirred at -10 0C for 2 h and filtered. Drying in vacuo at 25 CC for 66 h provided 85 g (49%) of product. The filtrate was concentrated in vacuo to a damp solid residue. This was taken up in 100 mL of methyl t-butyl ether. To the stirred suspension was added 300 mL of heptane and the resulting suspension was stirred at -10 0C for 2 h. The solid was filtered off, rinsed with 50 mL of methyl t-butyl etherheptane (1 :3) and pressed dry under suction. Further drying in vacuo at 34 °C for 6 h provided an additional 34.2 g (19.5%) of product. Total yield = 119.2 g (68.5%).
Step 5: Preparation of 2-(3-piperidin-4-ylidenemethyl-phenoxy)-5-trifluoromethyl-pyridine, hydrochloride
To a mixture of 4-[3-(5-trifluoromethyl-pyridin-2-yloxy)-benzylidene]-piperidine-1-carboxylic acid fert-butyl ester (312 g, 0.718 mol) in ethyl acetate (2.8 L) at 0 0C to -5 0C was added streamwise over a 20 min period, 4.0 M hydrogen chloride in dioxane (800 mL, 3.2 mol). No significant temperature change was noted. The resulting suspension was stirred at temperatures reaching 22 0C over the next 17 h. The suspension was filtered. The solid was washed with EtOAc (500 mL) and pressed as dry as possible under suction. The damp solid was dried in vacuo at 33 0C for 7 h to afford 225 g (84%) of product.
Step 6: Preparation of N-pyridin-3-yl-4-(3-(f5-(trifluoromethyl)pyridin-2-ylloxy}benzylidene)piperidine-1-carboxamide
To a mixture of 2-(3-piperidin-4-ylidenemethyl-phenoxy)-5-trifluoromethyl-pyridine (80.0 g, 0.216 mol) and phenyl pyridin-3-ylcarbamate (48.6 g, 0.227 mol) in acetonitrile (650 mL) was added dropwise diisopropylethyl amine (55.8 g, 0.432 mol). A solution formed after -45 min of stirring. The slightly turbid solution was stirred at ambient temperature for 18 h. TLC showed a prominent product spot with traces of both starting materials and two other fast moving spots. The solution was concentrated in vacuo to a viscous oil. This was partitioned between dichloromethane (600 mL) and water (500 mL). The aqueous layer was extracted with 200 mL of dichloromethane. The combined organic layers were washed with successive portions of 500 mL of 5% sodium hydroxide, and 200 mL of water, then dried over magnesium sulfate and concentrated in vacuo to 139.5 g of a viscous oil. This was dissolved in 350 ml_ of warm (50 0C) methyl t-butyl ether. Soon after a solution formed, solid began separating. The crystallizing mixture was kept at -10 0C for 4 h and filtered. The solid was rinsed with 60 ml_ of methyl t-butyl ether and pressed dry under suction. Further drying in vacuo at 28 0C for 16 h and then at 35 °C for 6 h provided 93.2 g (95%) of product.
PAPER
ACS Medicinal Chemistry Letters (2011), 2(2), 91-96
https://pubs.acs.org/doi/10.1021/ml100190t





PAPER
Science (Washington, DC, United States) (2017), 356(6342), 1084-1087
Pfizer Products Inc.WO2008047229
References
- ^ Johnson DS, Stiff C, Lazerwith SE, Kesten SR, Fay LK, Morris M, et al. (February 2011). “Discovery of PF-04457845: A Highly Potent, Orally Bioavailable, and Selective Urea FAAH Inhibitor”. ACS Medicinal Chemistry Letters. 2 (2): 91–96. doi:10.1021/ml100190t. PMC 3109749. PMID 21666860.
- ^ Jump up to:a b “JZP 150”. AdisInsight. 26 December 2023. Retrieved 16 August 2024.
- ^ “A Study of JZP150 in Adults With Posttraumatic Stress Disorder – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov.
- [1]. Johnson DS, et al. Discovery of PF-04457845: A Highly Potent, Orally Bioavailable, and Selective Urea FAAH Inhibitor. ACS Med Chem Lett. 2011 Feb 10;2(2):91-96. [Content Brief][2]. Ahn K, et al. Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J Pharmacol Exp Ther. 2011 Jul;338(1):114-24. [Content Brief][3]. Buntyn RW, et al. Inhibition of Endocannabinoid-Metabolizing Enzymes in Peripheral Tissues Following Developmental Chlorpyrifos Exposure in Rats. Int J Toxicol. 2017 Jan 1:1091581817725272. [Content Brief]
////////Redafamdastat, PF 04457845, JZP-150, JZP150, PF-04457845, PF-4457845, PF04457845, PF4457845, Q7119045



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com