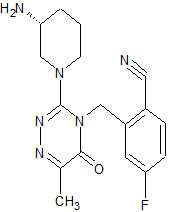

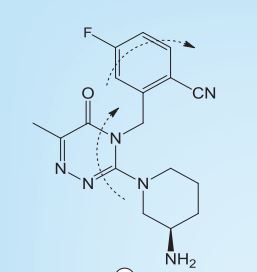
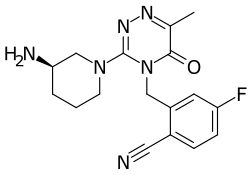
FOTAGLIPTIN
CAS 1312954-58-7
342.37, C17 H19 F N6 O
Benzonitrile, 2-[[3-[(3R)-3-amino-1-piperidinyl]-6-methyl-5-oxo-1,2,4-triazin-4(5H)-yl]methyl]-4-fluoro-
(R)-2-((3-(3-amino-piperidin-1-yl)-6-methyl-5-oxo-1,2,4-piperazine-4(5H)-yl)methyl)-4-fluorobenzonitrile,

BENZOATE cas 1403496-40-1 [ china 2024, approvals 2024 ]
(R) 2- Methyl-5-oxo-1,2,4-triazin-4 (5H) -yl) methyl) -4-fluorobenzonitrile (3- benzoate (compound benzoate A), of the formula: the C . 17 the H 19 the FN . 6 O · the C . 7 the H . 6 O 2 , molecular weight: 464.49.
useful as a dipeptidyl peptidase IV (DPPIV) inhibitor for treating diabetes, particularly type 2 diabetes
Dipeptidyl peptidase IV inhibitor,
a DPPIV inhibitor, being developed by Chongqing Fochon, with licensee Shenzhen Salubris Pharmaceuticals, for treating type 2 diabetes mellitus. In January 2017, fotagliptin benzoate was reported to be in phase 1 clinical development. The compound of the present invention was first disclosed in WO2011079778. See WO2015110078 and WO2015110077, claiming crystalline polymorphic form of the DPPIV inhibitor.
- Originator Chongqing Fochon Pharmaceutical
- Class Antihyperglycaemics
- Mechanism of Action CD26 antigen inhibitors
-
Shanghai Fosun Pharma Transfers Development Rights in New Diabetes & Cancer Therapies to Swiss-Greek Firm
Fotagliptin (SAL067) is a DPP-4 inhibitor under development for the treatment of type 2 diabetes. Like other DPP-4 inhibitors, it works by increasing endogenously produced GLP-1 and GIP.[1][2][3] In a phase 3 trial it showed similar results as alogliptin.[4]
Shanghai Fosun Pharma Transfers Development Rights in New Diabetes & Cancer Therapies to Swiss-Greek Firm
On 23 October 2013, leading Chinese healthcare company Shanghai Fosun Pharmaceutical Group Co., Ltd. signed an agreement with Sellas Life Science Group, a Switzerland based Greek pharmaceutical R&D company. According to the agreement, Fosun Pharma transfers to Sellas the global rights (excluding China) in development, commercialisation, marketing and distribution of Fotagliptin Benzoate and Pan-HER Inhibitors, two novel compounds owned by Fosun Pharma’s subsidiary Chongqing Fochon Pharmaceutical Co. Ltd.
Fotagliptin Benzoate is developed by Chongqing Fochon independently and has a prospect of developing into type 2 diabetes medicines, whereas Pan-HER Inhibitors, a receptor inhibitor of which Chongqing Fochon owns the proprietary IP rights, is a potential therapy for curing lung, breast and other cancers. Chongqing Fochon has filed application for international patent under the Patent Cooperation Treaty in respect of the two compounds.
The estimated total consideration for the transaction of approximately RMB3.248 billion will be paid by installment. In addition, upon the compounds obtaining relevant approvals in the US and/or Europe, Chongqing Fochon will be entitled to a 10% royalty in these regions on net revenue sales for eight years.
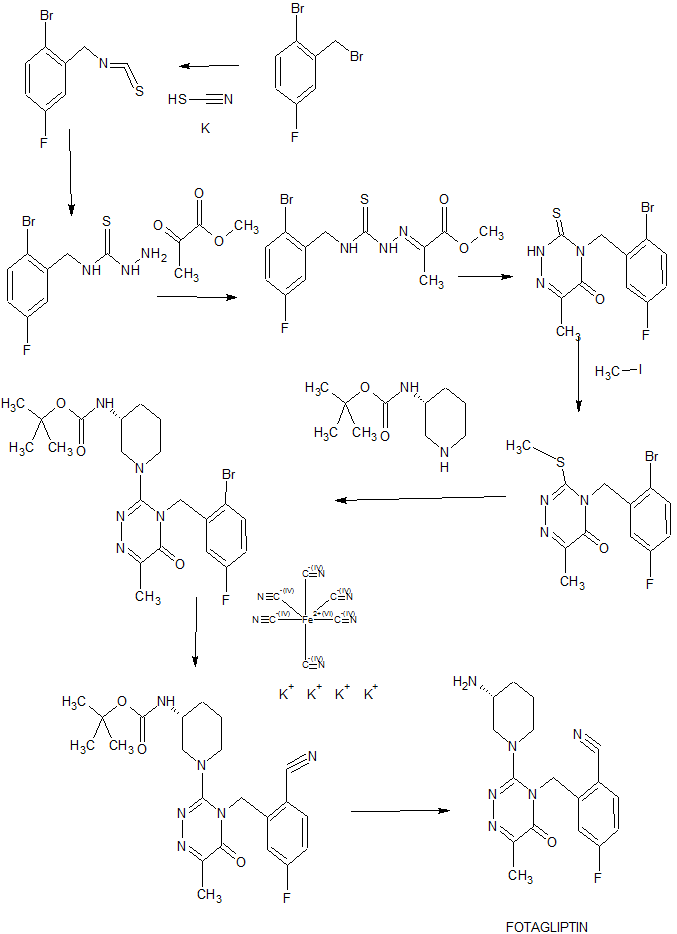
SYNTHESIS
Research Article
Development and validation of a UPLC–MS/MS method for simultaneous determination of fotagliptin and its two major metabolites in human plasma and urine
Zhenlei Wang1, Ji Jiang1, Pei Hu1 & Qian Zhao*,1
Aim: Fotagliptin is a novel dipeptidyl peptidase IV inhibitor under clinical development for the treatment of Type II diabetes mellitus. The objective of this study was to develop and validate a specific and sensitive ultra-performance liquid chromatography (UPLC)–MS/MS method for simultaneous determination of fotagliptin and its two major metabolites in human plasma and urine. Methodology & results: After being pretreated using an automatized procedure, the plasma and urine samples were separated and detected using a UPLC-ESI–MS/MS method, which was validated following the international guidelines. Conclusion: A selective and sensitive UPLC–MS/MS method was first developed and validated for quantifying fotagliptin and its metabolite in human plasma and urine. The method was successfully applied to support the clinical study of fotagliptin in Chinese healthy subjects.
PATENT
WO2011079778
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011079778&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription
PATENT
WO2015110078
compound A can be prepared according to the method disclosed in PCT / CN2010 / 080370, the specific synthesis route and the main reaction conditions are as follows:

Example 1 Preparation of 1-bromo-4-fluoro-2- (isothiocyanatomethyl) benzene (2)
To a DMF solution (20 ml) of 1-bromo-2- (bromomethyl) -4-fluorobenzene (1,5.36 g, 20.0 mmol) was added sodium iodide (1.20 g, 8.00 mmol) and potassium thiocyanate (3.88 g, 40.0 mmol). After the mixture was heated to 80C under nitrogen atmosphere for 12 hours, it was cooled to room temperature, 100 ml of water was added thereto, and extracted with ethyl acetate (50 mL x 2). The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, The concentrate was concentrated by suction to give a crude product, and the residue was purified by silica gel column chromatography (eluent: petroleum ether) to give 1-bromo-4-fluoro-2- (isothiocyanatomethyl) benzene (2).
Example 2 Preparation of N- (2-bromo-5-fluorobenzyl) hydrazinocarbothioamide (3)
A solution of hydrazine hydrate (80%, 2.22 g, 35.5 mmol) in 1,4-dioxane (20 mL) was cooled to 0 ° C and 1-bromo-4-fluoro-2- (isothiocyanate Yl) benzene (2,3.16 g, 12.8 mmol) in 1,4-dioxane (5 ml). The mixture was stirred at room temperature for 2 h, to which was added 100 ml of ice water, solid precipitated, filtered, washed with water and dried over phosphorus pentoxide overnight to give N- (2-bromo-5-fluorobenzyl) hydrazinothiocarb Amide (3).
MS: m / z, 278 (100%, M + 1), 280 (100%), 300 (10%, M + 23), 302 (10%).
Example 3 Preparation of methyl 2- (2- (2-bromo-5-fluorobenzylaminothioformamide) hydrazino) propionate (4)
N- (2-bromo-5-fluorobenzyl) hydrazinocarbothioamide (3, 1.12 g, 4.00 mmol) was added successively to a solution of pyruvic acid (352 mg, 4.00 mmol) in methanol And the residue was extracted with ethyl acetate (150 ml). The organic layer was washed successively with water, saturated sodium bicarbonate solution and saturated brine, and dried over anhydrous magnesium sulphate (MgSO4). The organic layer was washed with water, Dried, and concentrated by suction filtration to give methyl 2- (2- (2-bromo-5-fluorobenzylaminothioformamide) hydrazino) propionate (4).
MS: m / z, 362 (100%, M + 1), 364 (100%), 384 (60%, M + 23), 386 (60%).
Example 4 4- (2-Bromo-5-fluorobenzyl) -6-methyl-3-thioxo-3,4-dihydro-1,2,4-triazin- (5)
Sodium methoxide (0.4 M), freshly prepared from sodium (273 mg, 11.88 mmol) and dry methanol (30 ml), was dissolved in 30 ml of methanol, and methyl 2- (2- (2-bromo-5-fluorobenzylamino sulfide The mixture was heated to reflux for 22 h. Most of the solvent was distilled off. The residue was diluted with 100 ml of water, adjusted to pH = 1-2 with 2N concentrated hydrochloric acid, and the residue was extracted with ethyl acetate. The extract was washed with brine, dried over anhydrous sodium sulfate and concentrated by suction to give a crude product which was purified by silica gel column chromatography (eluent: ethyl acetate / petroleum ether = 20% -30%) to give 4- (2-bromo-5-fluorobenzyl) -6-methyl-3-thioxo-3,4-dihydro- ) -one (5).
MS: m / z, 330 (65%, M + 1), 332 (60%, M + 23).
Example 5 Preparation of 4- (2-bromo-5-fluorobenzyl) -6-methyl-3- (methylthio) -1,2,4-triazin-5 (4H) preparation
A mixture of 4- (2-bromo-5-fluorobenzyl) -6-methyl-3-thioxo-3,4-dihydro- , 914 mg, 2.77 mmol) was suspended in ethanol (15 ml), followed by addition of sodium hydroxide (111 mg, 2.77 mmol) and methyl iodide (787 mg, 5.54 mmol). The reaction mixture was diluted with 100 ml of water and extracted with ethyl acetate (30 ml x 2). The combined layers were washed with saturated brine, dried over anhydrous magnesium sulfate, concentrated by suction, and the residue was recrystallized from the residue. Silica gel column chromatography (eluent: ethyl acetate / petroleum ether = 20-25%) afforded 4- (2-bromo-5-fluorobenzyl) -6-methyl-3- (methylthio) -l, 2,4-triazin-5 (4H) -one (6).
1 the H NMR (400MHz, of DMSO, ppm by): [delta] 7.73 (m, IH), 7.16 (br, IH), 7.05 (D, IH), 5.09 (S, 2H), 2.56 (S, 3H), 2.32 ( S, 3H).
MS: m / z, 344 (100%, M + l), 346 (100%).
Example 6 (R) -tert-Butyl 1- (4- (2-bromo-5-fluorobenzyl) -6-methyl-5-oxo-4,5-dihydro- -triazin-3-yl) piperidine-3-carbamate (8)
A solution of 4- (2-bromo-5-fluorobenzyl) -6-methyl-3- (methylthio) -1,2,4-triazin-5 (4H) Mmol) and (R) -tert-butylpiperidine-3carbamate (7,208 mg, 1.04 mmol) for 5 min and heated to 135 ° C for 13 h under nitrogen. The reaction mixture was purified by column chromatography on silica gel (R) -tert-Butyl 1- (4- (2-bromo-5-fluorobenzyl) -6-methyl-5- Oxo-4,5-dihydro-1,2,4-triazin-3-yl) piperidine-3-carbamate (8).
MS: m / z, 496 (100%, M + l), 498 (100%).
Example 7 (R) -tert-Butyl 1- (4- (2-cyano-5-fluorobenzyl) -6-methyl-5-oxo-4,5-dihydro- Triazin-3-yl) piperidine-3-carbamate (9)
To a mixture of sodium carbonate (53 mg, 0.50 mmol), palladium acetate (3 mg, 0.013 mmol) and N-methylpyrrolidone 0.5 ml was added 3 drops of isopropanol and 2 drops of water, and the mixture was stirred at room temperature for 5 minutes, (R) -tert-Butyl 1- (4- (2-bromo-5-fluorobenzyl) -6-methyl-5-oxo-4,5-dihydro- – triazin-3-yl) piperidine-3-carbamate (8,246mg, 0.496mmol) in NMP (1.0mL), and heated to 140 ℃, then add the K 4 [of Fe (the CN) . 6 ] 3H · 2 O (209mg, 0.496 mmol), was heated at 140 ℃ 12h, cooled to room temperature, water was added 10ml, extracted with ethyl acetate (20mL × 2), the combined organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, (R) -tert-Butyl l- (4- (2-cyano-5- (2-fluoro-4-methoxyphenyl) Fluoro-benzyl) -6-methyl-5-oxo-4,5-dihydro-1,2,4-triazin-3-yl) piperidine-3-carbamate (9).
MS: m / z, 418 (20%), 443 (100%, M + 1), 465 (95%, M + 23).
Example 5 Preparation of compound A (R) -2 – ((3- (3-aminopiperidin- 1 -yl) -6-methyl- -yl) methyl) -4-fluorobenzonitrile (10)
To a solution of (R) -tert-Butyl 1- (4- (2-cyano-5-fluorobenzyl) -6-methyl-5-oxo-4,5-dihydro- Yl) piperidine-3-carbamate (9,37 mg) in 1 ml of methylene chloride was added 0.5 ml of trifluoroacetic acid and the mixture was stirred at room temperature for 1 hour, neutralized with a saturated sodium hydrogencarbonate solution, (Eluent: dichloromethane / methanol / aqueous ammonia = 92: 6: 2), in order to obtain (10ml × 3), the organic layer was dried over anhydrous sodium sulfate and concentrated in vacuo to give the crude product, which was purified by silica gel column chromatography Methyl) -5-oxo-1,2,4-triazin-4 (5H) -yl) methyl) -4-fluorobenzonitrile (10), i.e. Compound A.
1 the H NMR (400MHz, of DMSO, ppm by): [delta] 7.96 (m, IH), 7.36 (br, IH), 7.29 (D, IH), 5.23 (S, 2H), 3.15 (m, 3H), 2.72 ( 2H), 2.23 (s, 3H), 1.78 (d, 1H), 1.64 (d, 1H), 1.47 (m, 1H), 1.12 (m, 1H).
MS: m / z, 343 (100%, M + l).
Methyl-5-oxo-1,2,4-triazin-4 (5H) -yl) -2-oxoquinoline-3- Methyl) -4-fluorobenzonitrile benzoate (Compound A benzoate)
Configuration 95% ethanol solution: 500mL beaker by adding 228mL ethanol, add 12mL of water, stir well, spare.
60g of 95% ethanol, 120mL of 95% ethanol, stirring, dissolving, filtering, washing with 95% ethanol 18ml; to make the 500mL reaction flask, The ethanolic solution of benzoic acid was added dropwise at an internal temperature of 15 ° C. After completion of the dropwise addition, 95% ethanol was washed and dried under reduced pressure to constant weight to give 42.4 g of (R) -2- (3- (3-aminopiperidin-1-yl) -6-methyl- 1,2,4-triazin-4 (5H) -yl) methyl) -4-fluorobenzonitrile benzoate (the product).
Melting point determination: Instrument: Tianjin University Precision Instrument Factory YRT-3 melting point instrument.
Detection method: Take appropriate amount of this product, small study, 60 ° C, 2 hours of vacuum drying, according to the Chinese Pharmacopoeia 2010 edition two appendix Ⅵ C determination of the product melting point of 95 ℃ -115 ℃.
(5H) -benzoic acid was isolated from (R) -2- (3- (3-aminopiperidin-l- yl) -6-methyl- Methyl) -4-fluorobenzonitrile benzoate 0.1g, according to the Chinese Pharmacopoeia 2010 edition of two Appendix Ⅲ “General Identification Test” under the “benzoate” test method for testing, set 10ml volumetric flask, Add water and dilute the solvent to the mark, shake, the precise amount of 5ml to 10ml beaker, adjust the solution of phenolphthalein was neutral, drop of ferric chloride solution, were observed ocher precipitation. At the same time do blank control test, the results: multiple batches of samples of benzoic acid identification test results were positive, reagent blank does not interfere with the determination of specificity.
Identification HPLC: chromatographic conditions for the introduction of the Eclipse Plus C the Agilent 18 column (5μm, 4.6х250mm), detection wavelength of 229nm, mobile phase of acetonitrile: 0.1% phosphoric acid = 7: 3, a flow rate of 1.0ml / min, The injection volume was 20μl.
The compound A (7.5 mg) of Example 8 was dissolved in a 50 mL volumetric flask, diluted with 70% aqueous acetonitrile and diluted to the mark, shaken as a solution of the compound A reference substance; and 12.5 mg of benzoic acid in a 25 mL volumetric flask, With a volume ratio of 70% acetonitrile aqueous solution and diluted to the mark, take 1mL in 25mL volumetric flask, with volume ratio of 70% acetonitrile aqueous solution and diluted to the mark, shake, as benzoic acid reference substance solution; take this product 10mg In a 50mL volumetric flask, with a volume ratio of 70% acetonitrile aqueous solution dissolved and diluted to the mark, shake, as the product A benzoic acid salt of the test solution. Respectively, the precise amount of the reference solution and the test solution 20μl, according to high performance liquid chromatography (Chinese Pharmacopoeia 2010 edition two Appendix VD), according to the chromatographic conditions of injection, chromatogram shown in Figure 1, Method.
The results showed that the retention time of the main peak was the same as the retention time of the reference substance, and the content of compound A and benzoic acid was calculated by the peak area. The molar ratio of compound A and benzoic acid was 1: 1.
Infrared absorption spectrum identification: the United States NICOLET AVATAR 330FT-IR infrared spectrometer, in accordance with the Chinese Pharmacopoeia 2010 edition two Appendix IVC correction, take the amount of goods, using KBr tablet method for determination of the product of the infrared diffraction pattern (Figure 2 shown) to wave number cm & lt -1 , he said in 3419.75cm -1 , 2936.46cm -1 , 2230.38cm -1 , 1683.28cm -1 , 1609.47cm -1 , 1511.65cm -1 , 1419.44cm -1 , 829.18cm -1 , 722.67cm -1 characteristic absorption peak, 0.2cm error is ± -1 .
NEW PATENT
WO-2017008684
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017008684&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=FullText
Shenzhen Salubris Pharmaceuticals Co Ltd, α-Crystal form of compound A, preparation method thereof, and pharmaceutical composition comprising same
Dipeptidyl peptidase IV (DPP-IV) is a serine protease that specifically hydrolyzes the N-terminal Xaa-Pro or Xaa-Ala dipeptide of a polypeptide or protein. DPP-IV is an atypical serine protease whose Ser-Asp-His catalytic triad at the C-terminal region is different from a typical serine protease in reverse order.
DPP-IV has a variety of physiologically relevant substrates, such as inflammatory chemokines, normal T-cell expressed and secreted (RANTES), eotaxin and macrophage Cell-derived chemokines, neuropeptides such as neuropeptide Y (NPY) and P5 substances, vasoactive peptides, incretin such as glucagon-like peptide-1 (GLP-1) And glucose-dependent insulinotropic polypeptide (GIP).
Inhibition of DPP-IV in vivo resulted in increased levels of endogenous GLP-1 (7-36) and decreased production of its antagonist GLP-1 (9-36). Thus, DPP-IV inhibitors may be effective in diseases associated with DPP-IV activity such as type 2 diabetes, diabetic dyslipidemia, impaired Glucose Tolerance (IGT), impaired Fasting Plasma Glucose (IFG ), Metabolic acidosis, ketosis, appetite regulation and obesity.
DPP-IV inhibitor Alogliptin (Alogliptin) clinically for type 2 diabetes showed good therapeutic effect, approved in the United States market. Therefore, DPP-IV inhibitors are currently considered to be novel therapeutic approaches for the treatment of type 2 diabetes
PCT / CN2010 / 080370 describes a series of DPP-IV inhibitors with neo-nuclear structure. (R) -2 – ((3- (3-aminopiperidin- 1 -yl) -6-methyl-5-oxo-l, 2,4- tris piperazine -4 (5H) – yl) methyl) -4-fluorobenzonitrile (using the prior art process to obtain the product as a yellow oil), molecular formula: the C . 17 the H 19 the FN . 6 O, molecular weight: 342 chemical formula The following formula (I)
In order to improve the medicinal properties of the compound, studies with favorable stability properties can be effectively used in the treatment of patients with pathological conditions by inhibiting DPP-IV in pharmaceutical compositions.
It is an object of the present invention to provide a stable crystalline form of a stable competitive inhibitor compound D of a reversible dipeptidyl peptidase-IV (DPP-IV).
The chemical name of compound A is: (R) -2 – ((3- (3-aminopiperidin- 1 -yl) -6-methyl-5-oxo-1,2,4-triazin- 5H) – yl) methyl) -4-fluorobenzonitrile, molecular formula: the C . 17 the H 19 the FN . 6 O, molecular weight: 342, the chemical structure of formula a compound of the following formula (the I),
compound A can be prepared according to the method disclosed in PCT / CN2010 / 080370, the specific synthesis route and the main reaction conditions are as follows:
EXAMPLE 1 Preparation of Compound A.
Compounds A were prepared according to the procedures of PCT / CN2010 / 080370 Examples 2 and 3 using the following synthetic route:
The resulting compound of the A, 1 the H-NMR (400MHz, of DMSO, ppm by): [delta] 7.96 (m, IH), 7.36 (br, IH), 7.29 (D, IH), 5.23 (S, 2H), 3.15 (m, 3H), 2.72 (m, 2H), 2.23 (s, 3H), 1.78 (d, 1H), 1.64 (d, , 343 (100%, M + l).
Specific preparation steps are as follows:
Step A. 1-bromo-4-fluoro-2- (isothiocyanatomethyl) benzene (2)
To a DMF solution (20 mL) of 1-bromo-2- (bromomethyl) -4-fluorobenzene (1,5.36 g, 20.0 mmol) was added sodium iodide (1.20 g, 8.00 mmol) and potassium thiocyanate (3.88 g, 40.0 mmol). The mixture was heated to 80 ° C under nitrogen atmosphere for 12 hours, cooled to room temperature, and 100 mL of water was added thereto. The mixture was extracted with ethyl acetate (50 mL × 2). The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, The concentrate was concentrated by suction to give a crude product, and the residue was purified by silica gel column chromatography (eluent: petroleum ether) to give 1-bromo-4-fluoro-2- (isothiocyanatomethyl) benzene (2).
Step BN- (2-Bromo-5-fluorobenzyl) hydrazinocarbothioamide (3)
Dioxane solution (20 mL) of hydrazine hydrate (80%, 2.22 g, 35.5 mmol) was cooled to 0 ° C, and thereto was added 1-bromo-4-fluoro-2- (isothiocyanate Yl) benzene (2,3.16 g, 12.8 mmol) in 1,4-dioxane (5 mL). The mixture was stirred at room temperature for 2 h, and 100 mL of ice water was added thereto. The solid was precipitated, filtered, washed with water and dried over phosphorus pentoxide overnight to give N- (2-bromo-5-fluorobenzyl) hydrazinothiazepine Amide (3). MS: m / z, 278 (100%, M + 1), 280 (100%), 300 (10%, M + 23), 302 (10%).
Step C. Methyl 2- (2- (2-bromo-5-fluorobenzylaminothiocarboxamide) hydrazino) propanoate (4)
N- (2-bromo-5-fluorobenzyl) hydrazinocarbothioamide (3, 1.12 g, 4.00 mmol) was added successively to a solution of pyruvic acid (352 mg, 4.00 mmol) in methanol And the residue was extracted with ethyl acetate (150 mL). The organic layer was washed successively with water, saturated sodium hydrogencarbonate solution and saturated brine, and dried over anhydrous magnesium sulphate (MgSO4). The organic layer was washed with water, Dried and concentrated by suction filtration to give methyl 2- (2- (2-bromo-5-fluorobenzylaminothioformamide) hydrazino) propionate (4). MS: m / z, 362 (100%, M + 1), 364 (100%), 384 (60%, M + 23), 386 (60%).
Step D. 4- (2-Bromo-5-fluorobenzyl) -6-methyl-3-thioxo-3,4-dihydro-1,2,4-triazin- (4)
Sodium methoxide (0.4 M), freshly prepared from sodium (273 mg, 11.88 mmol) and dry methanol (30 mL), was dissolved in 30 mL of methanol and methyl 2- (2- (2-bromo-5-fluorobenzylamino sulfide The mixture was heated to reflux for 22 h. Most of the solvent was distilled off. The residue was diluted with 100 mL of water and the pH was adjusted to 1 to 2 with concentrated hydrochloric acid (2N). The solvent was evaporated under reduced pressure. The extract was washed with brine, dried over anhydrous sodium sulfate and concentrated by suction to give a crude product which was purified by silica gel column chromatography (eluent: ethyl acetate / petroleum ether = 20% 4- (2-bromo-5-fluorobenzyl) -6-methyl-3-thioxo-3,4-dihydro-1,2,4-triazin-5 (2H ) -one (5), MS: m / z, 330 (65%, M + 1), 332 (60%, M + 23).
(4H) -one (6) & lt; EMI ID = 36.1 & gt; [0161] Step 4. 4- (2-Bromo-5-fluorobenzyl) -6 -methyl-
Methyl-3-thioxo-3,4-dihydro-1,2,4-triazin-5 (2H) -one (5,914 (111 mg, 2.77 mmol) and methyl iodide (787 mg, 5.54 mmol) were added successively to 15 mL of ethanol. The reaction mixture was diluted with 100 mL of water and extracted with ethyl acetate (30 mL × 2). The combined layers were washed with saturated brine, dried over anhydrous magnesium sulfate, concentrated by suction filtration, and the residue was recrystallized from the residue. (2-bromo-5-fluorobenzyl) -6-methyl-3- (methylthio) – (2-bromo-5-fluorobenzyl) -2-methylbenzene was purified by silica gel column chromatography (eluent: ethyl acetate / petroleum ether = 20-25% 1,2,4-triazine -5 (4H) – one (. 6). 1 the H NMR (400MHz, of DMSO, ppm by): [delta] 7.73 (m, IH), 7.16 (br, IH), 7.05 (D, 1H), 5.09 (s, 2H), 2.56 (s, 3H), 2.32 (s, 3H). MS: m / z, 344 (100%, M + 1), 346 (100%).
Step F. Preparation of (R) -tert-Butyl 1- (4- (2-bromo-5-fluorobenzyl) -6-methyl-5-oxo-4,5-dihydro- – three -3-yl) piperidin-3-ylcarbamate (8)
A solution of 4- (2-bromo-5-fluorobenzyl) -6-methyl-3- (methylthio) -1,2,4-triazin-5 (4H) -one (6,180 mg, 0.523 mmol ) And (R) -tert-butylpiperidine-3-carbamate (7, 208 mg, 1.04 mmol) for 5 min and heated to 135 ° C under nitrogen for 13 h. The reaction mixture was purified by silica gel column chromatography (R) -tert-Butyl 1- (4- (2-bromo-5-fluorobenzyl) -6-methyl-5-oxo-propan-1- (8). MS: m / z, 496 (100%, M + l), 498 (M + l) (100%).
Step G. Preparation of (R) -tert-Butyl 1- (4- (2-cyano-5-fluorobenzyl) -6-methyl-5-oxo-4,5-dihydro- – three -3-yl) piperidine-3-carbamate (9)
To a mixture of sodium carbonate (53 mg, 0.50 mmol), palladium acetate (3 mg, 0.013 mmol) and 0.5 mL of N-methylpyrrolidone was added 3 drops of isopropanol and 2 drops of water, and the mixture was stirred at room temperature for 5 minutes, (R) -tert-Butyl 1- (4- (2-bromo-5-fluorobenzyl) -6-methyl-5-oxo-4,5-dihydro- 3-yl) piperidine-3-carbamate (8,246mg, 0.496mmol) in NMP (1.0mL), and heated to 140 ℃, then add the K 4 [of Fe (the CN) . 6 ] .3H 2 O (209 mg, 0.496 mmol), heated at 140 ° C for 12 h, cooled to room temperature, and 10 mL of water was added thereto. The mixture was extracted with ethyl acetate (20 mL × 2). The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate and concentrated by suction filtration to give (R) -tert-Butyl 1- (4- (2-cyano-5-fluorobenzyl) – (2-cyano-5-fluorophenyl) -carbamic acid ethyl ester 6-methyl-5-oxo-4,5-dihydro-1,2,4-triazin-3-yl) piperidine-3- carbamate (9). MS: m / z, 418 (20%), 443 (100%, M + 1), 465 (95%, M + 23).
Methyl-5-oxo-1,2,4-triazin-4 (5H) -ylidene-2-methyl- ) methyl ) -4-fluorobenzonitrile (10, compound A)
To a solution of (R) -tert-Butyl 1- (4- (2-cyano-5-fluorobenzyl) -6-methyl-5-oxo-4,5-dihydro- Yl) piperidine-3-carbamate (9,37 mg) in dichloromethane was added 0.5 mL of trifluoroacetic acid and the mixture was stirred at room temperature for 1 hour, neutralized with saturated sodium hydrogencarbonate solution, (Eluent: dichloromethane / methanol / aqueous ammonia = 92: 6: 2) to obtain (R (10mL × 3), the combined organic layer was dried over anhydrous sodium sulfate and concentrated in vacuo to give a crude product, which was purified by silica gel column chromatography Methyl-5-oxo-1,2,4-triazin-4 (5H) -yl) methyl) – 2- Fluorobenzonitrile (10 as a yellow oil).
1 the H NMR (400MHz, of DMSO, ppm by): [delta] 7.96 (m, IH), 7.36 (br, IH), 7.29 (D, IH), 5.23 (S, 2H), 3.15 (m, 3H), 2.72 ( (M, 2H), 2.23 (s, 3H), 1.78 (d, 1H), 1.64 (d, 1H), 1.47 , M + 1).
Patent
CN 104803972
REFERENCES
CN 104803972
CN 104803971
US 20110160212
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This article is a compilation for educational purposes only.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
References
- Wu, Min; Li, Qian-Qian; Zhang, Hong; Zhu, Xiao-Xue; Li, Xiao-Jiao; Li, Ying; Sun, Hai-Gang; Ding, Yan-Hua (June 2021). “Safety, Pharmacokinetics, and Pharmacodynamics of a Dipeptidyl Peptidase-4 Inhibitor: A Randomized, Double-Blinded, Placebo-Controlled Daily Administration of Fotagliptin Benzoate for 14 Days for Type 2 Diabetes Mellitus”. Clinical Pharmacology in Drug Development. 10 (6): 660–668. doi:10.1002/cpdd.895. ISSN 2160-763X. PMID 33440080. S2CID 231606176.
- Fang, Lan; Gao, Zhenguo; Wu, Songgu; Jia, Shengzhe; Wang, Jingkang; Rohani, Sohrab; Gong, Junbo (1 August 2021). “Ultrasound-assisted solution crystallization of fotagliptin benzoate: Process intensification and crystal product optimization”. Ultrasonics Sonochemistry. 76 105634. Bibcode:2021UltS…7605634F. doi:10.1016/j.ultsonch.2021.105634. ISSN 1350-4177. PMC 8261672. PMID 34218067.
- Ding, Yanhua; Zhang, Hong; Li, Cuiyun; Zheng, WenBo; Wang, Meng; Li, Ying; Sun, HaiGang; Wu, Min (3 June 2021). “Safety and pharmacokinetic interaction between fotagliptin, a dipeptidyl peptidase-4 inhibitor, and metformin in healthy subjects”. Expert Opinion on Drug Metabolism & Toxicology. 17 (6): 725–731. doi:10.1080/17425255.2021.1915283. PMID 33899649. S2CID 233400233.
- Xu, Mingtong; Sun, Kan; Xu, Wenjie; Wang, Chuan; Yan, Dewen; Li, Shu; Cong, Li; Pi, Yinzhen; Song, Weihong; Sun, Qingyuan; Xiao, Rijun; Peng, Weixia; Wang, Jianping; Peng, Hui; Zhang, Yawei; Duan, Peng; Zhang, Meiying; Liu, Jianying; Huang, Qingmei; Li, Xuefeng; Bao, Yan; Zeng, Tianshu; Wang, Kun; Qin, Li; Wu, Chaoming; Deng, Chunying; Huang, Chenghu; Yan, Shuang; Zhang, Wei; Li, Meizi; Sun, Li; Wang, Yanjun; Li, HongMei; Wang, Guang; Pang, Shuguang; Zheng, Xianling; Wang, Haifang; Wang, Fujun; Su, Xiuhai; Ma, Yujin; Zhang, Wei; Li, Ziling; Xie, Zuoling; Xu, Ning; Ni, Lin; Zhang, Li; Deng, Xiangqun; Pan, Tianrong; Dong, Qijuan; Wu, Xiaohong; Shen, Xingping; Zhang, Xin; Zou, Qijing; Jiang, Chengxia; Xi, Jue; Ma, Jianhua; Sun, Jingchao; Yan, Li (2023). “Fotagliptin monotherapy with alogliptin as an active comparator in patients with uncontrolled type 2 diabetes mellitus: a randomized, multicenter, double-blind, placebo-controlled, phase 3 trial”. BMC Medicine. 21 (1): 388. doi:10.1186/s12916-023-03089-x. ISSN 1741-7015. PMC 10563289. PMID 37814306.
//////////FOTAGLIPTIN BENZOATE, FOTAGLIPTIN , PHASE 1, 1403496-40-1, 1312954-58-7
N[C@@H]1CCCN(C1)C3=NN=C(C)C(=O)N3Cc2cc(F)ccc2C#N
















Maybe you should try agarosebased chromatography. Why did you chose Silica?