















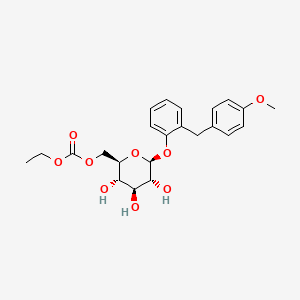
Sergliflozin Etabonate
408504-26-7 Cas no
Ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-[2-[(4-methoxyphenyl)methyl]phenoxy]oxan-2-yl]methyl carbonate
2-(4-methoxybenzyl)phenyl 6-O-ethoxycarbonyl-beta-D-glucopyranoside
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-[2-[(4-methoxyphenyl)methyl]phenoxy]tetrahydropyran-2-yl]methyl carbonate
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-{2-[(4-methoxyphenyl)methyl]phenoxy}oxan-2-yl]methyl carbonate
PHASE 2……….TYPE 3 DIABETES AND OBESITY
A SGLT-2 inhibitor potentially for the treatment of type 2 diabetes and obesity.
GW-869682; GW-869682X; KGT-1251
- etabonate de sergliflozine
- etabonato de sergliflozina
MW 448.4, C23H28O9
KISSEI INNOVATOR
GSK DEVELOPER
Sergliflozin Etabonate is a benzylphenol glucoside and selective sodium-glucose co-transporter subtype 2 (SGLT2) inhibitor with antihyperglycemic activity. Its prodrug form, sergliflozin etabonate, is orally available and is converted to sergiflozin upon absorption.
Sergliflozin etabonate (INN/USAN,[1][2] codenamed GW869682X) is an investigational anti-diabetic drug being developed by GlaxoSmithKline. It did not undergo further development after phase II
Sergliflozin inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine.[3][4]
Chemistry
Etabonate refers to the ethyl carbonate group. The remaining structure, which is the active substance, is called sergliflozin.

Sergliflozin
[PDF] Design, Syntheses, and SAR Studies of Carbocyclic Analogues of …onlinelibrary.wiley.com974 × 740Search by imageDesign, Syntheses, and SAR Studies of Carbocyclic Analogues of Sergliflozin as Potent SodiumDependent Glucose Cotransporter 2 In
Sergliflozin Etabonate is a benzylphenol glucoside and selective sodium-glucose co-transporter subtype 2 (SGLT2) inhibitor with antihyperglycemic activity. Its prodrug form, sergliflozin etabonate, is orally available and is converted to sergiflozin upon absorption.

sergliflozin and prodrugs of sergliflozin, in particular sergliflozin etabonate, including hydrates and solvates thereof, and crystalline forms thereof. Methods for its manufacture are described in the patent applications EP 1344780 and EP 1489089 for example.
The compounds are described in EP 1 329 456 A1 and a crystalline form ofSergliflozin etabonate is described in EP 1 489 089 A1.
PATENT
US6872706B2
https://patentscope.wipo.int/search/en/detail.jsf?docId=US40677423&_cid=P20-MF4ZUQ-42384-1
PATENT
WO2001068660A1
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2001068660&_cid=P20-MF4ZXC-45172-1
SYN
Heterocycles 2016, 92, 1599
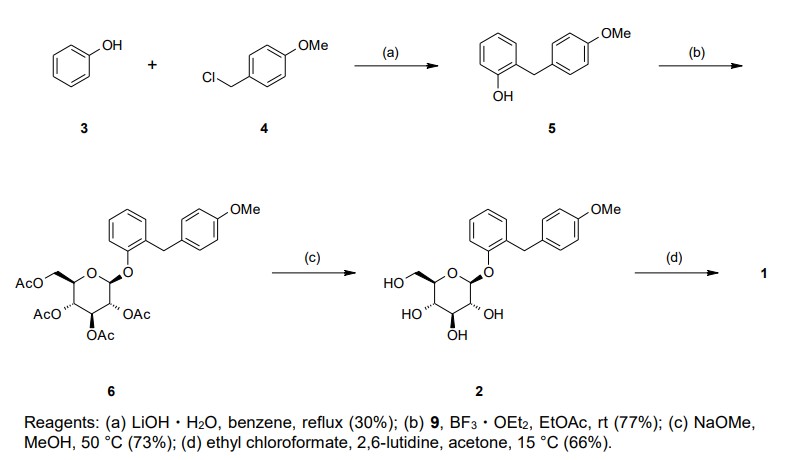
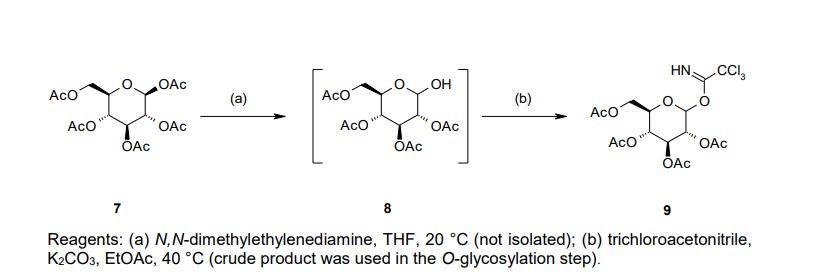
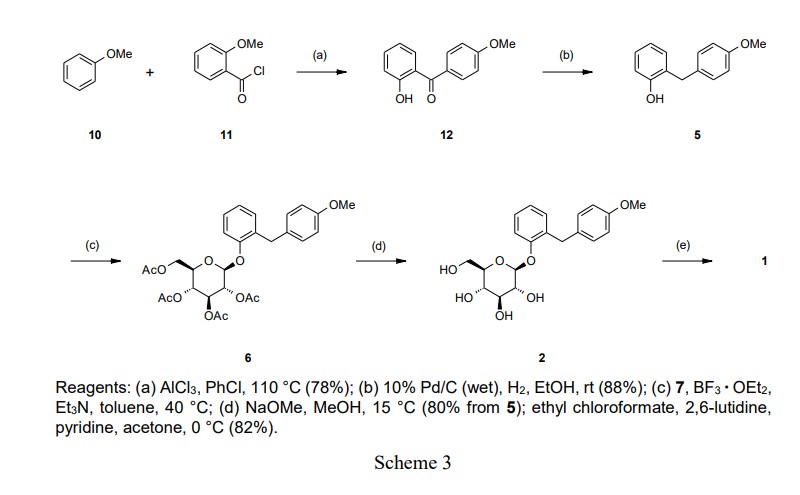
Our initial synthetic route of Serglifrozin etabonate (1) in early development consisted of six steps,
including synthesis of tetra-O-acetyl-D-glucopyranosyl trichloroacetimidate (9), as shown in Scheme 1
and Scheme 2 The first step is the coupling reaction of phenol (3) and 4-methoxybenzyl chloride (4) in the presence of
lithium hydroxide monohydrate (LiOH·H2O) to provide the aglycon 5 in a 30% yield following
chromatographic purification (Scheme 1). We prepared 9 separately by mono-deacetylation of
penta-O-acetyl-β-D-glucopyranose (7) with N,N-dimethylethylenediamine in THF followed by reaction of
the crude product of 8 with trichloroacetonitrile in the presence of potassium carbonate (K2CO3) in ethyl
acetate (EtOAc) (Scheme 2). Next, we carried out glycosylation of 5 with 9 in the presence of boron
trifluoride diethyl etherate (BF3·OEt2) in EtOAc to produce 6 in a 77% yield. The obtained 6 was
deacetylated with sodium methoxide (NaOMe) in MeOH to produce Serglifrozin (2) in a 73% yield, and
reaction of the isolated 2 with ethyl chloroformate in the presence of 2,6-lutidine in acetone provided 1 in
a 66% yield. The overall yield from 3 was 11%. While this route was capable of supplying small
amounts of 1, it suffered from several disadvantages.
The coupling reaction between 3 and 4 provided the aglycon 5 in low yield (30%); thus, chromatographic
purification was required to obtain highly pure 5. The trichloroimidation reaction of 8 is too hazardous
for large-scale manufacturing, because an excess amount of trichloroacetonitrile, a volatile and highly
toxic reagent, is required to obtain the trichloroacetimidate 9. Furthermore, 9 is too unstable to use
conveniently in large-scale manufacturing. Trichloroacetamide, a sublimation compound, is formed as a
by-product from the glycosylation of 5 with 9. Thus, the vacuum line and the vacuum pump of the
manufacturing equipment would be polluted by trichloroacetamide.
Because of these issues, this synthetic method is unsuitable for large-scale manufacturing. Therefore,
we investigated alternative processes for the preparation of 1, suitable for large-scale manufacturing. An improved synthetic method for 1 was achieved in a five-step procedure without purification of 6
(intermediate), as shown in Scheme 3.
The Friedel-Crafts acylation of anisole (10) with 2-methoxybenzoyl chloride (11) in the presence of
aluminum chloride (AlCl3) at 110 °C provided benzophenone (12), which was selectively demethylated
on the methoxy group at the 2-position. The crude product of 12 was crystallized from MeOH to
provide highly pure 12 in a 78% yield. Hydrogenation of 12 in EtOH with 0.3–0.4 MPa H2 at room
temperature in the presence of 10% Pd/C provided 5. The crude product of 5 was crystallized from
toluene/n-heptane to provide highly pure 5 in an 88% yield.
The key step of the synthesis was the formation of the O-glycosylated product 6. In the initial synthesis,
it was necessary to isolate 6 to remove trichloroacetamide. Consequently, 2 was provided in a 56%
yield from 5. To obtain 6 efficiently without using the trichloroacetimidate (9), we evaluated several
conditions for the direct O-glycosylation of 5 with 7. The results are summarized in Table 1. The
O-glycosylation of 5 with 7 (200 mol%) in the presence of boron trifluoride diethyl etherate (BF3·OEt2;
100 mol%) in dichloromethane (DCM) at room temperature provided the crude product of 6 with a good
yield (80%) and β-selectivity (94/6), and then the deacetylation of the crude product of 6 in the presence
of sodium methoxide (NaOMe) in MeOH proceeded almost quantitatively to provide 2 in a 71% isolated
yield from 5 (run 1). Using this method, it was not necessary to isolate 6 because the excess amount of
7 was converted to glucose and removed to the aqueous layers in the deacetylation step. Use of DCM is
undesirable for large-scale manufacturing because quenching of O-glycosylation with water is highly
exothermic and washing of the DCM layer with water is a complicated procedure. Additionally, it is
strongly desirable to avoid using DCM in a manufacturing process due to environmental issues. For the reasons mentioned above, we attempted to use toluene as an alternative solvent. The O-glycosylation in
the presence of BF3·OEt2 (100 mol%) in toluene at 30 °C did not proceed completely, and the yield of 6
was lower than run 1 (run 2). We concluded that the lower solubility of 7 in toluene, compared with
DCM, caused the low yield. Because it was difficult to increase the amount of toluene from the
perspective of manufacturing efficiency, we tried to improve its solubility by optimizing the reagent
equivalent. Fortunately, we found that an excess amount of BF3·OEt2 enhanced the solubility of 7 in
toluene, and using 300 mol% of BF3·OEt2 in toluene provided 6 in a good yield (80%), similar to that
when using DCM (run 3). In contrast, reducing the amount of 7 provided 6 in an insufficient yield, and
2 was consequently provided in a lower yield (60%) (run 4). To achieve higher β-selectivity and an
increased yield, triethylamine (Et3N) was added to the O-glycosylation of 5 with 7 in the presence of
BF3·OEt2, according to the method of Lee et al.
9 Addition of Et3N (30 mol%) at 30 °C resulted in both
higher yield (89%) and higher β-selectivity (97/3) to provide 2 with a 79% isolated yield (run 5).
Increasing the amount of Et3N to 60 mol% at 30 °C resulted in a lower yield (85%) of 6 compared with
run 5, and the yield of 2 decreased (74%) (run 6). Increasing the reaction temperature to 40 °C in the
presence of 60 mol% of Et3N achieved the best results for both high yield (90%) and high β-selectivity
(99/1) to provide 2 in an 80% yield (run 7).
6-O-Ethoxycarbonyl-2-[(4-methoxyphenyl)methyl]phenyl-β-D-glucopyranoside (1). Ethyl
chloroformate (407 mg, 3.75 mol) was added drop-wise to the mixture of 2 (1.13 g, 3.0 mmol) and
2,6-lutidine (563 mg, 5.25 mmol) in acetone (4 mL) while maintaining the temperature between 12 and
18 °C. The reaction mixture was stirred at 15 °C for 23 h. Water (5 mL) was added drop-wise while
maintaining the temperature below 30 °C, and EtOAc (10 mL) was then added to the mixture. The
biphasic solution was transferred to a separating funnel for phase separation. The aqueous layer was
extracted with EtOAc (5 mL). The EtOAc layers were combined, washed successively with an aqueous
solution of 10% citric acid (5 mL × 2), an aqueous solution of 10% NaCl (5 mL), an aqueous solution of
5% NaHCO3 (5 mL × 2), and an aqueous solution of 10% NaCl (5 mL). They were then dried over
Na2SO4 and the filtrate was concentrated under reduced pressure. EtOH was added to the residue, and
the weight was adjusted to 7.2 g. The mixture was heated to 65 °C to dissolve solids. The solution was
cooled to 55 °C and seeded with 1. The solution was aged for 1 h at 50 °C, during which time the
product began to crystallize. After the slurry was cooled to 25 °C, n-heptane (11 mL) was added
drop-wise to the slurry at 25 °C followed by stirring for 1 h at 25 °C. The slurry was cooled to 3 °C and
then stirred for 2 h at 3 °C. The slurry was filtered, and the wet cake was washed with a mixed solvent
of EtOH (1.5 mL) and n-heptane (3 mL). The precipitate was dried in vacuo at 70 °C to give 888 mg
(66% yield) of 1 as a white solid. [α]
20
D -43.5 (c 1.0, DMSO). IR (KBr) cm-1
: 3495, 1744, 1514, 1488,
1454, 1467, 1411, 1372, 1340, 1266. 1H-NMR (CDCl3) δ: 1.27 (3H, t, J=7.0 Hz), 2.00 (1H, d, J=1.6
Hz), 3.46–3.54 (3H, m), 3.56–3.61 (2H, m), 3.72 (1H, d, J=2.1 Hz), 3.75 (3H, s), 3.82 (1H, d, J=15.5 Hz),
4.03 (1H, d, J=15.5 Hz), 4.11–4.22 (2H, m), 4.42 (2H, d, J=3.8 Hz), 4.69 (1H, d, J=7.4 Hz), 6.79–6.83
(2H, m), 6.97–7.02 (2H, m), 7.04–7.07 (2H, m), 7.15–7.22 (2H, m). 13C-NMR (CDCl3) δ: 14.2 (q), 36.1
(t), 55.4 (q), 64.4 (t), 66.4 (t), 69.6 (d), 73.4 (d), 73.8 (d), 75.7 (d), 100.8 (d), 114.1 (d×2), 114.4 (d), 122.7
(d), 128.0 (d), 129.2 (d×2), 130.0 (s), 131.1 (d), 133.4 (s), 155.2 (s), 155.4 (s), 157.8 (s). HRMS (ESI)
m/z: 466.2070 [M+NH4]
+
(Calcd for C23H32NO9: 466.2072)
6-O-Ethoxycarbonyl-2-[(4-methoxyphenyl)methyl]phenyl-β-D-glucopyranoside (1). Ethyl
chloroformate (21.6 g, 0.199 mol) was added drop-wise to the mixture of 2 (65.0 g, 0.173 mol),
2,6-lutidine (27.8 g, 0.259 mol) and pyridine (0.33 g, 4.2 mmol) in acetone (210 mL), maintaining the
temperature between -1 and 5 °C. The reaction mixture was stirred at 0 °C for 2 h. The reaction was
monitored by HPLC.15 Water (200 mL) was added drop-wise, maintaining the temperature below 30 °C,
and then EtOAc (220 mL) was added to the mixture. The biphasic solution was transferred to a
separating funnel for phase separation. The aqueous layer was extracted with EtOAc (140 mL). The
EtOAc layers were combined, washed successively with an aqueous solution of 10% citric acid (180 mL
× 2), an aqueous solution of 10% NaCl (66 g), an aqueous solution of 5% NaHCO3 (65 g × 2), and an aqueous solution of 10% NaCl (100 g), and then dried over Na2SO4 (65 g). After acetic acid (10 g,
0.167 mol) was added to the filtrate, the mixture was concentrated under reduced pressure. The residue
was dissolved in EtOH (660 mL) at 65 °C. The solution was concentrated under reduced pressure until
more than 330 mL distillate had been collected. EtOH was added to the residue, and the weight was
adjusted to 370 g. n-Heptane (120 mL) was added, and the resulting slurry was heated to 65 °C to
dissolve solids. The solution was cooled to 55 °C and seeded with 1. The solution was aged for 1 h at
50 °C, during which time the product began to crystallize. n-Heptane (480 mL) was added drop-wise to
the slurry, maintaining the temperature between 50 and 60 °C, and the slurry was stirred for 0.5 h at 55 °C.
The slurry was allowed to cool slowly over 2.5 h to 30 °C, then cooled to 3 °C, and then stirred for 1.5 h
at 3 °C. The slurry was filtered, and the wet cake was washed with a mixed solvent of EtOH (80 mL)
and n-heptane (180 mL). The precipitate was dried in vacuo at 70 °C to give 63.6 g (82% yield) of 1 as
a white solid.
REFERENCES (AND NOTES)
- W. N. Washburn, Expert Opin. Ther. Patents, 2009, 19, 1485.
- A. M. Pajor and E. M. Wright, J. Biol. Chem., 1992, 267, 3557.
- E. M. Wright, Am. J. Physiol. Renal Physiol., 2001, 280, F10.
- Y. Kanai, W. S. Lee, G. You, D. Brown, and M. A. Hediger, J. Clin. Invest., 1994, 93, 397.
- H. Fujikura, N. Fushimi, T. Nishimura, K. Tatani, and M. Isaji, PCT, WO 02/28872 (2002).
- H. Fujikura, N. Fushimi, T. Nishimura, K. Tatani, K. Katsuno, M. Hiratochi, Y. Tokutake, and M.
Isaji, PCT, WO 01/688660 (2001). - K. Katsuno, Y. Fujimori, Y. Takemura, M. Hiratochi, F. Itoh, Y. Komatsu, H. Fujikura, and M. Isaji,
J. Pharmacol. Exp. Ther., 2007, 320, 323. - M. Isaji, Curr. Opin. Investig. Drugs, 2007, 8, 285.
- S. Y. Lee, S. E. Rho, K. Y. Min, T. B. Kim, and H. K. Kim, J. Carbohydr. Chem., 2001, 20, 503.
- M. Yamaguchi, A. Horiguchi, A. Fukuda, and T. Minami, J. Chem. Soc., Perkin Trans. 1, 1990,
1079. - K. Ishihara, H. Kurihara, and H. Yamamoto, J. Org. Chem., 1993, 58, 3791.
- I. T. Akimova, A. V. Kaminsky, and V. I. Svistunova, Chem. Heterocycl. Compd., 2005, 41, 1374.
- B. N. Cook, S. Bhakta, T. Biegel, K. G. Bowman, J. I. Armstrong, S. Hemmerich, and C. R. Bertozzi,
J. Am. Chem. Soc., 2000, 122, 8612. - HPLC conditions: column, Inertsil ODS-3 (5 µm) 4.6 mm × 250 mm (GL Science Inc.); mobile
phase, isocratic elution with acetonitrile / 0.02 M KH2PO4, pH 3 = 6/4; flow rate, 1.0 mL/min;
column oven temperature, 40 °C; wave length, 225 nm; retention times, 5 = 16 min, α-anomer of 5 =18 min. - HPLC conditions: column, Inertsil ODS-3 (5 µm) 4.6 mm × 250 mm (GL Science Inc.); mobile
phase, gradient elution with 5 min 4/6 → 15 min 6/4 → 30 min 6/4 of acetonitrile/0.02 M KH2PO4,
pH 3; flow rate, 1.0 mL/min; column oven temperature, 40 °C; wavelength, 225 nm; retention times,
1 = 17 min, 2,6- and 4,6-bis-O-ethoxycarbonyl derivatives = 24 min, 3,6-bis-O-ethoxycarbonyl
derivative = 25 min.
SYN
Synthesis 2024, 56, 906–943
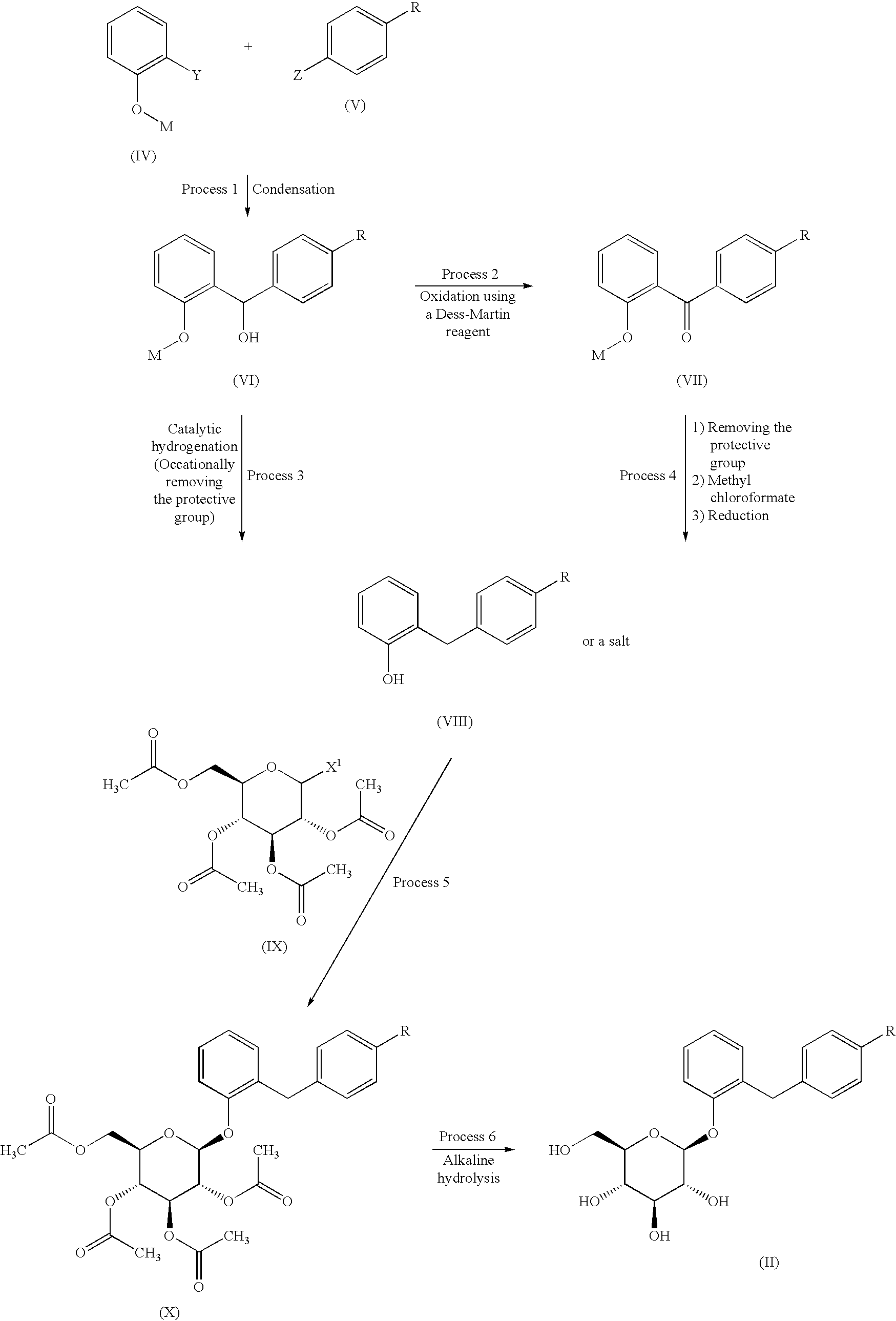
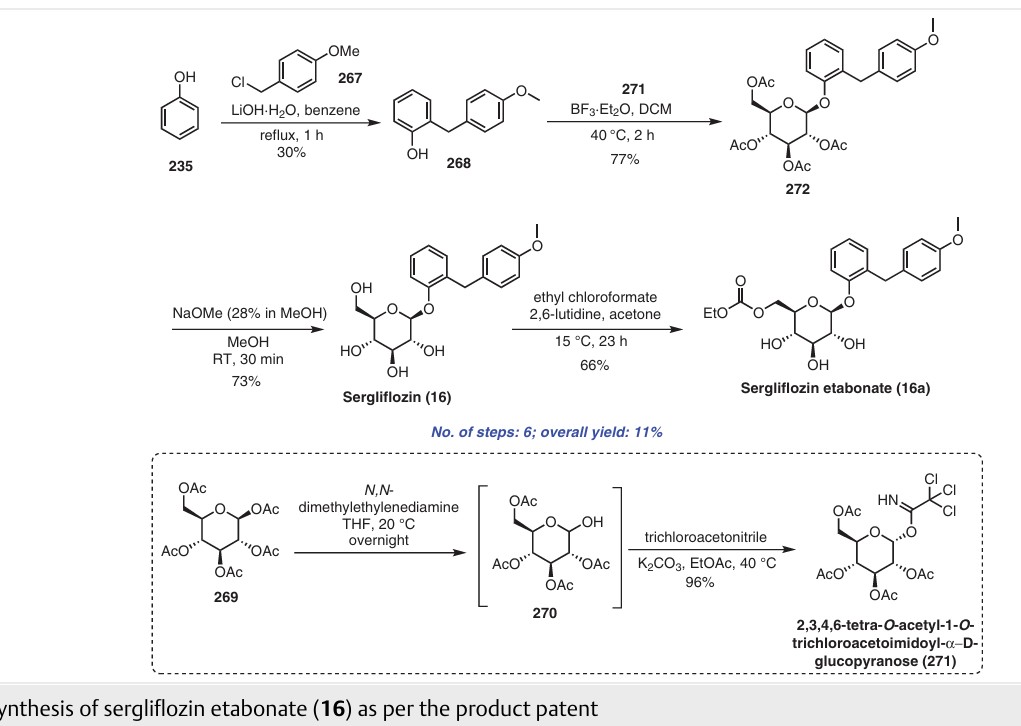
Sergliflozin etabonate (16), also known as GW869682X, was developed collaboratively by GlaxoSmithKline and Kissei Pharmaceutical (Japan). Unfortunately, it did not pass phase III trials. It belongs to the class of sodium–glucose linked transporter 2 (SGLT2) inhibitors and acts as a prodrug of sergliflozin, with the ethyl carbonate group referred to as etabonate. When compared to phlorizin, sergliflozin etabonate demonstrated significantly higher activity against SGLT2 than SGLT1. The initial synthetic route for the preparation of sergliflozin was described and patented by Kissei Pharmaceutical Co., Ltd. This particular route for Oaryl-glycoside-type derivatives was registered in the United States under patent application number US6872706B2.73 The first reported synthesis of sergliflozin etabonate
(16), which involves six steps, can be found in the patents US6872706B2 73a and WO2001068660A1 (Scheme 48).73b Compound 271 was prepared in a high yield of 96% follow ing a literature procedure. The selective monodeacetylationof penta-O-acetyl-b-D-glucopyranose, compound 269, was
achieved using N,N-dimethylethylenediamine in THF, resulting in the formation of compound 270. Subsequently, a reaction with trichloroacetonitrile and potassium carbonate led to the synthesis of intermediate 271 in excellent yield. To prepare the aglycone intermediate 268, phenol (235) was condensed with 4-methoxybenzyl chloride (267) using LiOH under reflux conditions. Further,O-glycosyla
tion of compound 268 with 271 was accomplished using boron trifluoride–diethyl etherate (BF3·OEt2), yielding intermediate 272. Removal of the acetyl groups from intermediate 272 was carried out using NaOMe in methanol to obtain sergliflozin (16a) in a yield of 73%. Finally, sergliflozin etabonate (16) was obtained by reacting compound 16a with ethyl chloroformate and 2,6-lutidine, resulting in a yield of
66%. The overall yield of sergliflozin etabonate (16a) was calculated to be 11%. It is important to note that the trichloroimidation reaction used in the synthesis of trichloroacetimidate 271 is considered hazardous and is not recommended for commercial use due to the highly toxic reagent, trichloroaceto
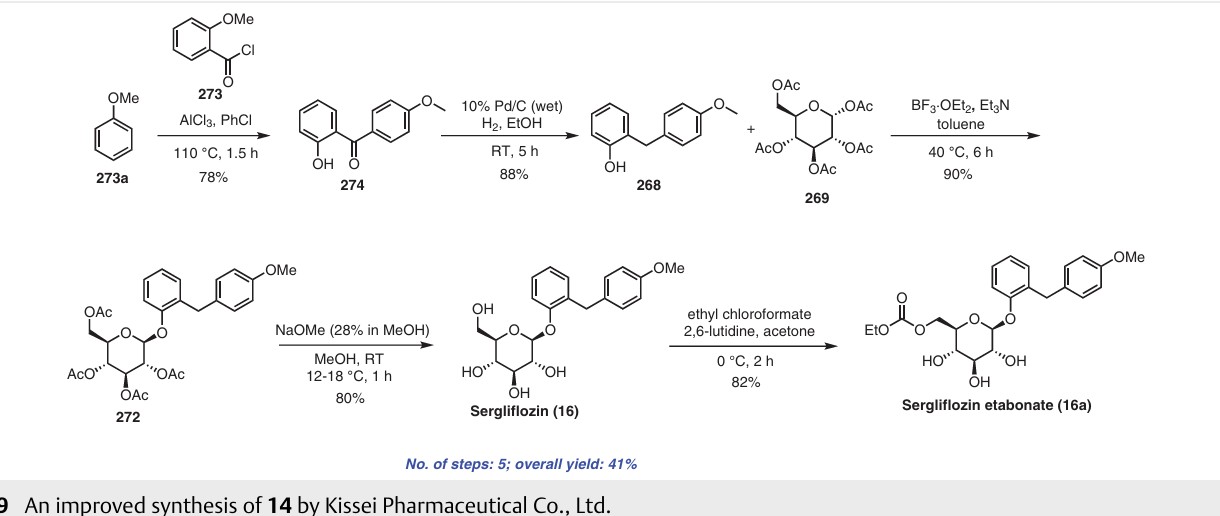
nitrile. Additionally, the process poses challenges in effectively removing the unwanted by-product, trichloroacetamide, formed during the preparation.A recently published approach presents an alternative synthesis of sergliflozin etabonate (16) that avoids the use of a trichloroacetimidate intermediate (Scheme 49).74a The five-step synthesis of compound 16a commenced from
readily available anisole (273a). An efficient Friedel–Crafts reaction was performed on anisole (273a) using the acid chloride 273 in the presence of aluminum chloride in chlorobenzene, leading to formation of benzophenone 274. Notably, demethylation of 274 was also observed under these
conditions. Next, ketone group reduction was achieved us ing 10% Pd/C and ethanol under 0.3–0.4 MPa of H2, providing compound 268 in 88% yield and high purity. Subsequently, O-glycosylation of 268 with penta-acetylated com pound 269 was carried out using BF3·Et2O and triethylamine, resulting in the formation of 272 in 90% yield with a high b-selectivity (99:1).74b Deacetylation of compound 272 was performed using NaOMe in methanol, affording sergliflozin (16a) in 80% yield. Further reaction with
ethyl chloroformate in the presence of 2,6-lutidine resulted in sergliflozin etabonate (16). The overall yield of compound 16 was calculated to be 41%. This novel synthetic route offers a promising alternative to the traditional method and demonstrates improved efficiency in the preparation of sergliflozin etabonate (16)
(73) (a) Fujikura, H.; Fushimi, N. US6872706B2, 2005. (b) Fujikura, H.; Fushimi, N.; Nishimura, T.; Tatani, K.; Katsuno, K.; Hiratochi, M.; Tokutake, Y.; Isaji, M. WO2001068660A1, 2001.
(74) (a) Kobayashi, M.; Isawa, H.; Sonehara, J.; Kubota, M. Heterocycles 2016, 92, 1599. (b) Lee, Y. S.; Rho, S. E.; Min, K. Y.; Kim, T. B.; Kim, H. K. J. Carbohydr. Chem. 2001, 20, 503.
| Patent | Submitted | Granted |
|---|---|---|
| Progression Inhibitor For Disease Attributed To Abnormal Accumulation Of Liver Fat [US2008045466] | 2008-02-21 | |
| NOVEL SUBSTITUTED TETRAHYDRONAPHTHALENES, PROCESS FOR THE PREPARATION THEREOF AND THE USE THEREOF AS MEDICAMENTS [US2010249097] | 2010-09-30 | |
| (CARBOXYLALKYLENEPHENYL)PHENYLOXAMIDES, METHOD FOR THE PRODUCTION THEREOF AND USE OF SAME AS A MEDICAMENT [US2010261645] | 2010-10-14 | |
| (CYCLOPROPYLPHENYL)PHENYLOXAMIDES, METHOD FOR THE PRODUCTION THEREOF, AND USE OF SAME AS A MEDICAMENT [US8148375] | 2010-10-14 | 2012-04-03 |
| Crystals of glucopyranosyloxybenzyl benzene derivative [US7371730] | 2005-06-02 | 2008-05-13 |
| CERTAIN CRYSTALLINE DIPHENYLAZETIDINONE HYDRATES, PHARMACEUTICAL COMPOSITIONS THEREOF AND METHODS FOR THEIR USE [US8003636] | 2009-08-13 | 2011-08-23 |
| NOVEL DIPHENYLAZETIDINONE SUBSTITUTED BY PIPERAZINE-1-SULFONIC ACID AND HAVING IMPROVED PHARMACOLOGICAL PROPERTIES [US2009264402] | 2009-10-22 | |
| Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, process for preparing them, medicaments comprising these compounds, and their use [US7759366] | 2009-08-27 | 2010-07-20 |
| Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same [US2005065098] | 2005-03-24 | |
| Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same [US6872706] | 2004-01-29 | 2005-03-29 |
| Patent | Submitted | Granted |
|---|---|---|
| PROGRESSION INHIBITOR FOR DISEASE ATTRIBUTED TO ABNORMAL ACCUMULATION OF LIVER FAT [US2009286751] | 2009-11-19 | |
| THERAPEUTIC USES OF SGLT2 INHIBITORS [US2011077212] | 2011-03-31 | |
| PHARMACEUTICAL COMPOSITION COMPRISING A SGLT2 INHIBITOR IN COMBINATION WITH A DPP-IV INHIBITOR [US2011098240] | 2011-04-28 | |
| Substituted imidazoline-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011112097] | 2011-05-12 | |
| Heterocycle-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising them and use thereof [US2011046105] | 2011-02-24 | |
| Arylchalcogenoarylalkyl-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011046185] | 2011-02-24 | |
| Arylchalcogenoarylalkyl-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011053947] | 2011-03-03 | |
| Novel aromatic fluoroglycoside derivatives, pharmaceuticals comprising said compounds and the use thereof [US2011059910] | 2011-03-10 | |
| Novel phenyl-substituted imidazolidines, process for preparation thereof, medicaments comprising said compounds and use thereof [US2011178134] | 2011-07-21 | |
| HETEROCYCLIC COMPOUNDS, PROCESSES FOR THEIR PREPARATION, MEDICAMENTS COMPRISING THESE COMPOUNDS, AND THE USE THEREOF [US2011183998] | 2011-07-28 |
| Systematic (IUPAC) name | |
|---|---|
| 2-(4-methoxybenzyl)phenyl 6-O-(ethoxycarbonyl)-β-D-glucopyranoside | |
| Clinical data | |
| Routes of administration | Oral |
| Identifiers | |
| CAS Number | 408504-26-7 |
| ATC code | None |
| PubChem | CID: 9824918 |
| IUPHAR/BPS | 4587 |
| ChemSpider | 21234810 |
| ChEMBL | CHEMBL450044 |
| Chemical data | |
| Formula | C23H28O9 |
| Molecular mass | 448.463 g/mol |
References
- World Health Organization (2008). “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 59” (PDF). WHO Drug Information. 22 (1): 66. Archived from the original (PDF) on February 19, 2009.
- “Statement on a nonproprietary name adopted by the USAN council: Sergliflozin etabonate” (PDF). American Medical Association. Retrieved 2008-08-10.
- Katsuno K, Fujimori Y, Takemura Y, et al. (January 2007). “Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level”. J Pharmacol Exp Ther. 320 (1): 323–30. doi:10.1124/jpet.106.110296. PMID 17050778. S2CID 8306408.
- “Prous Science: Molecule of the Month November 2007”. Archived from the original on 2007-11-05. Retrieved 2008-10-28.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
////////// etabonate, Sergliflozin etabonate, Sergliflozin, PHASE 3, GW869682X, GSK, KISSEI, GW-869682; GW-869682X; KGT-1251
CCOC(=O)OCC1C(C(C(C(O1)OC2=CC=CC=C2CC3=CC=C(C=C3)OC)O)O)O
CCOC(=O)OCC1C(C(C(C(O1)Oc2ccccc2Cc3ccc(cc3)OC)O)O)O