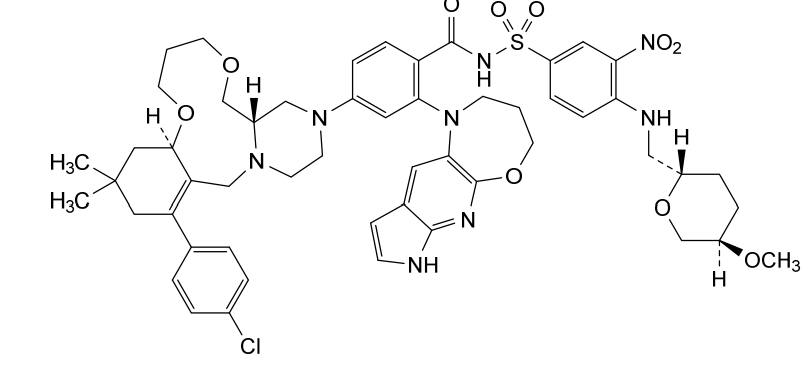
Surzetoclax, also known as ABBV 453; is a highly potent and selective BCL-2 inhibitor with a Ki of approximately 0.07 nM. It induces apoptosis in BCL-2–dependent hematologic cancer cells, showing EC50 values typically below 10 nM in sensitive models. In vivo, Surzetoclax causes rapid tumor regression in xenograft models of non-Hodgkin lymphoma (NHL) and chronic lymphocytic leukemia (CLL). It is orally bioavailable and demonstrates dose-dependent target engagement with favorable pharmacokinetics. Compared to Venetoclax, Surzetoclax was designed to reduce risks of tumor lysis syndrome and other dose-limiting toxicities.
Surzetoclax is a small molecule drug. The usage of the INN stem ‘-toclax’ in the name indicates that Surzetoclax is a B-cell lymphoma 2 (Bcl-2) inhibitor. Surzetoclax has a monoisotopic molecular weight of 1038.41 Da.
A Study to Assess Adverse Events and Change in Disease Activity of Oral ABBV-453 Alone or in Combination With Subcutaneous and/or Oral Antimyeloma Agents in Adult Participants With Multiple Myeloma (MM)CTID: NCT06953960Phase: Phase 1/Phase 2Status: RecruitingDate: 2025-12-01
A Study to Assess the Adverse Events and Change in Disease Activity in Adult Participants With Relapsed or Refractory Multiple Myeloma Receiving Oral ABBV-453 TabletsCTID: NCT05308654Phase: Phase 1Status: Active, not recruitingDate: 2025-08-14
A Study Assessing Adverse Event and How Oral ABBV-453 Moves Through the Body in Adult Participants With Relapsed or Refractory (R/R) Chronic Lymphocytic Leukemia (CLL)/Small Lymphocytic Lymphoma (SLL)CTID: NCT06291220Phase: Phase 1Status: Active, not recruitingDate: 2025-06-06
SYN
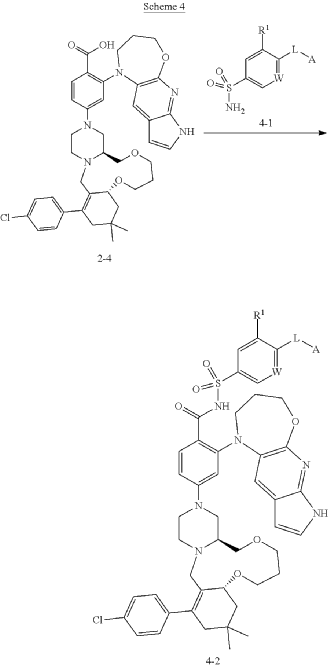
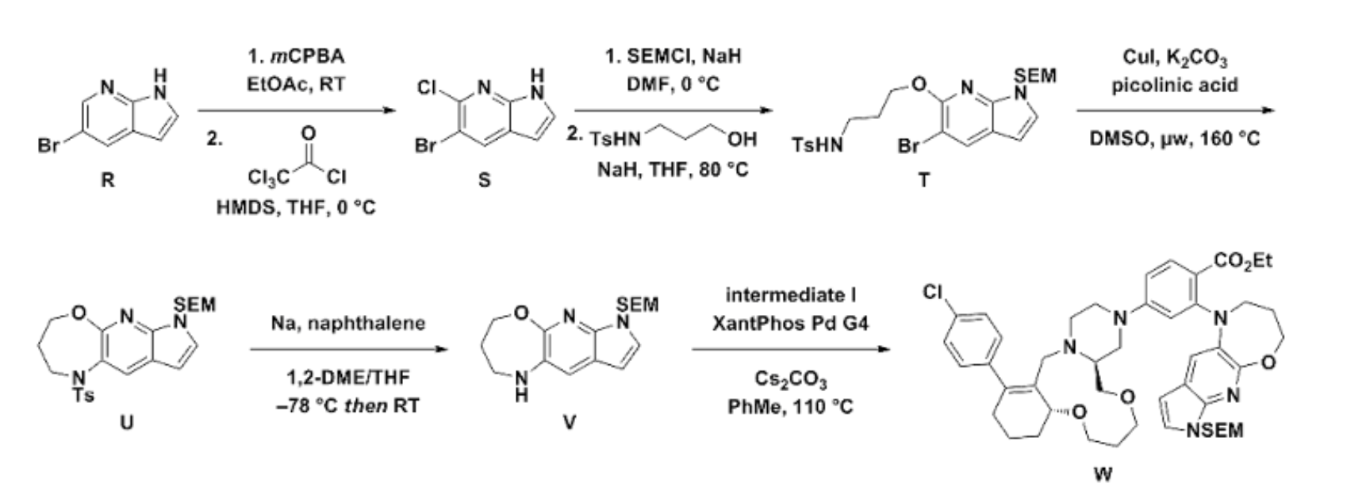
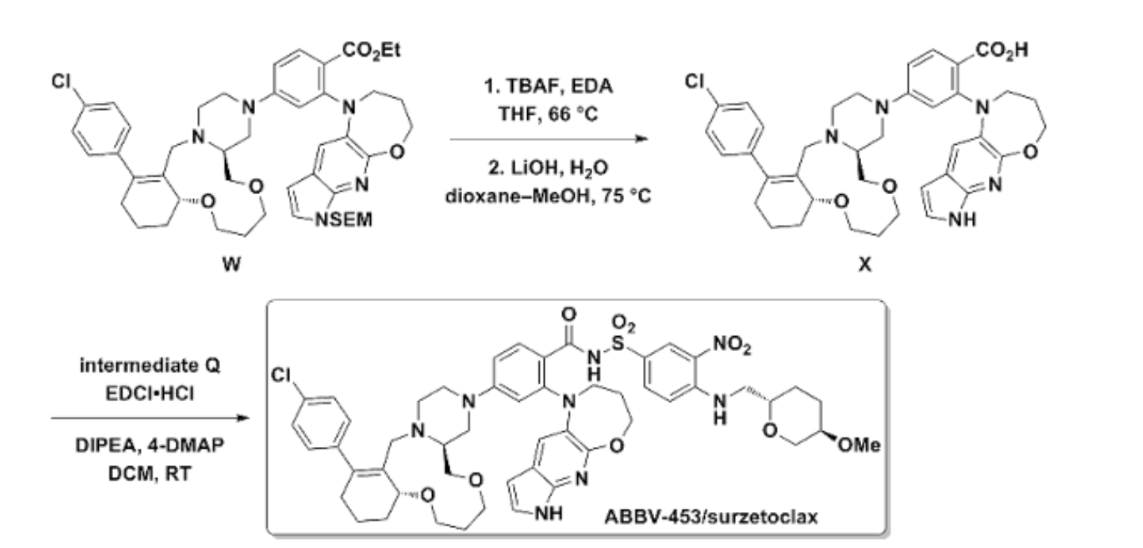
The synthesis of surzetoclax (ABBV-453), a complex, next-generation BCL-2 inhibitor, can be accomplished through a patented 27-step convergent route or a more streamlined, AI-assisted method that involves the modular assembly of three key fragments.
Patented Synthesis (Literature Route)
The published synthesis (described in patent WO 2023/141536 A1) is a 27-step convergent route with a 12-step longest linear sequence. The molecule is assembled from three main components: a 6,11,6-fused tricyclic core, a trans-1,2-disubstituted tetrahydropyran (THP) unit, and a 5,6,7-fused heteroaromatic system.
Key Steps and Intermediates:
Core Tricycle Assembly: The 6,11,6-fused macrocycle is formed by a sequence initiated from dimedone, involving a macrocyclization step using an 11-membered ring bis-triflate intermediate.
THP Fragment Construction: The THP moiety’s synthesis includes an enzymatic resolution step using porcine pancreatic lipase to establish the required stereochemistry.
Heteroaromatic System: The 7-azaindole-oxazepane tricycle is formed via a microwave-assisted, copper-mediated cyclization reaction.
Final Coupling: The fragments are joined through Buchwald–Hartwig and N-sulfonylamide coupling reactions to yield the final surzetoclax molecule.
AI-Assisted Synthesis (ChemAIRS Route)
A more efficient, human-directed AI retrosynthesis approach developed by Chemical.AI offers a more convergent and experimentally practical alternative. This route also uses three fragments but employs different, more efficient coupling strategies.
Key Features of the Revised Route:
Fragment 25a (Core Tricycle): Assembled in 10 steps using more accessible starting materials and a Mizoroki-Heck coupling to incorporate an aryl group.
Fragment 12a (THP Motif): The synthesis is streamlined to four steps from simple building blocks, utilizing a Mitsunobu reaction to introduce the azide precursor instead of the patent’s longer tosylation/displacement sequence.
Fragment 12b (Azaindole-Oxazepane Tricycle): An efficient nickel-photoredox C–N coupling is used to form an intermediate that then undergoes a base-promoted cyclization.
Final Assembly: Fragments 12a and 12b are joined using a palladium-catalyzed amidocarbonylation, followed by deprotection and a final SNAr coupling with fragment 25a to form surzetoclax.
The AI-assisted route achieves greater modularity and adaptability for potential scale-up compared to the patented process.
To a flask containing racemic (3,4-dihydro-2H-pyran-2-yl)methanol (25.8) was added dichloromethane (250 mL) and N,N-dimethylpyridin-4-amine (32.0 g). The flask was placed in an ice bath. After 15 minutes, neat acetyl chloride (16.1 mL) was added, and the reaction was stirred in the ice bath for 1 hour. The reaction mixture was poured into a separatory funnel containing water (200 mL) and the two layers were separated. The aqueous layer was extracted once with dichloromethane (80 mL). The combined organic layers were dried over magnesium sulfate and filtered. The filtrate was concentrated. The residue was purified using flash chromatography (330 g silica column, 0-60% ethyl acetate/heptanes) to afford racemic (3,4-dihydro-2H-pyran-2-yl)methyl acetate. To a flask containing water (1 L) was added potassium phosphate tribasic (4.48 g) and potassium dihydrogen phosphate (4.28 g) and the mixture was stirred until all the solids dissolved. To the solution was added racemic (3,4-dihydro-2H-pyran-2-yl)methyl acetate (10.0 g) in acetone (10 mL) at ambient temperature followed by porcine pancreatic lipase (75 mg). In another flask, the same reaction was set up on twice the scale. The reactions were stirred at ambient temperature for 18 hours then both were combined into a separatory funnel and extracted with 1:1 ethyl acetate/heptanes (2-500 mL). The combined organic layers were discarded, and the aqueous layer was extracted with 9:1 ethyl acetate/heptanes (2×500 mL). The combined organic layers were dried over magnesium sulfate and filtered. The filtrate was concentrated. The residue was purified using flash chromatography (80 g silica column, 0-100% ethyl acetate/heptanes) to afford the title compound. 1H NMR (500 MHz, CDCl 3) δ ppm 6.39 (dt, J=6.3, 2.0 Hz, 1H), 4.71 (dddd, J=6.3, 5.0, 2.5, 1.3 Hz, 1H), 3.96-3.88 (m, 1H), 3.76-3.62 (m, 2H), 2.12 (dddt, J=17.3, 10.8, 6.6, 2.4 Hz, 1H), 2.04-1.94 (m, 1H), 1.88 (dd, J=7.3, 5.4 Hz, 1H), 1.82-1.76 (m, 1H), 1.74-1.66 (m, 1H). A small amount of the material was treated with 3-(4-(trifluoromethyl)phenyl)propanoic acid, N,N-dimethylpyridin-4-amine and N 1-((ethylimino)methylene)-N 3,N 3-dimethylpropane-1,3-diamine hydrochloride to obtain (S)-(3,4-dihydro-2H-pyran-2-yl)methyl 3-(4-(trifluoromethyl)phenyl)propanoate. Analytical chiral supercritical fluid chromatography (ChiralPak AD-H, 5-50% CH 3OH, 3 mL/minute, 10 minutes method, 150 bar CO 2) was used to determine the ee of the ester to be 93%. Major enantiomer retention time was 1.26 minutes and that of the minor enantiomer was 1.34 minutes.
Example 25B
(S)-2-((benzyloxy)methyl)-3,4-dihydro-2H-pyran
To a flask containing sodium hydride (60 weight % in mineral oil, 1.104 g) was added tetrahydrofuran (60.0 mL). The slurry was cooled in an ice bath and a solution of Example 25A (2.100 g) in tetrahydrofuran (3 mL) was added dropwise followed by tetrahydrofuran (2 mL). The reaction was stirred in the ice bath for 15 minutes. Neat (bromomethyl)benzene (3.50 mL) was added, and the ice bath was removed. The reaction was stirred for 18 hours at ambient temperature. The reaction mixture was placed in an ice bath and carefully quenched with dropwise addition of 1:1 water/aqueous saturated ammonium chloride solution (80 mL). The biphasic mixture was extracted with 3:1 ethyl acetate/heptanes (2×50 mL). The combined organic layers were dried over magnesium sulfate, filtered and the filtrate was concentrated. The residue was purified using flash chromatography (80 g silica column, 0-5% ethyl acetate/heptanes) to afford the title compound. 1H NMR (500 MHz, CDCl 3) δ ppm 7.40-7.27 (m, 5H), 6.40 (dt, J=6.2, 2.0 Hz, 1H), 4.69 (dddd, J=6.3, 4.9, 2.5, 1.3 Hz, 1H), 4.62 (d, J=12.2 Hz, 1H), 4.58 (d, J=12.2 Hz, 1H), 4.03 (dddd, J=10.4, 6.4, 4.3, 2.3 Hz, 1H), 3.60 (dd, J=10.2, 6.3 Hz, 1H), 3.53 (dd, J=10.2, 4.3 Hz, 1H), 2.10 (dddt, J=17.2, 10.6, 6.5, 2.4 Hz, 1H), 2.02-1.92 (m, 1H), 1.90-1.81 (m, 1H), 1.70 (dtd, J=13.5, 10.4, 5.9 Hz, 1H).
To a solution of Example 25B (3.60 g) in tetrahydrofuran (50.0 mL) was added dropwise 9-borabicyclo[3.3.1]nonane (0.5 M in tetrahydrofuran, 75.0 mL) at 0° C. over 1 hour. The mixture was then stirred at ambient temperature for 18 hours. The reaction mixture was placed in an ice bath and 10% aqueous sodium hydroxide solution (25 mL) was carefully added to the mixture at 0° C., followed by 30% aqueous hydrogen peroxide solution (32 mL). The mixture was stirred at ambient temperature for 1 hour. The reaction mixture was quenched with saturated aqueous sodium sulfite solution (40 mL) at 0° C. and concentrated under reduced pressure to remove most of the organic solvent. The residue was extracted with 3:1 ethyl acetate/heptanes (2×50 mL). The combined organic layers were dried over magnesium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified using flash chromatography (120 g silica column, 0-60% ethyl acetate/heptanes) to afford the title compound. 1H NMR (500 MHz, CDCl 3) δ ppm 7.42-7.27 (m, 5H), 4.59 (d, J=12.2 Hz, 1H), 4.54 (d, J=12.2 Hz, 1H), 4.05 (ddd, J=10.8, 4.9, 2.3 Hz, 1H), 3.71 (td, J=10.2, 5.0 Hz, 1H), 3.53-3.38 (m, 3H), 3.14 (dd, J=10.8, 10.2 Hz, 1H), 2.14 (dtd, J=8.2, 4.7, 4.0, 2.3 Hz, 1H), 1.76-1.66 (m, 1H), 1.51-1.35 (m, 3H). LC/MS (APCI+) m/z 223.53 (M+H) +.
A solution of Example 25C (1.000 g) in tetrahydrofuran (20.0 mL) was cooled in an ice bath. Solid sodium hydride (60 weight % in mineral oil, 0.270 g) was added and the mixture was stirred in the ice bath for 15 minutes. Neat iodomethane (0.400 mL) was added and the reaction was allowed to warm to ambient temperature and stirred for 16 hours. The reaction was quenched by adding 0.1% aqueous solution of trifluoroacetic acid (4 mL) and water (20 mL), then extracted 5:1 ethyl acetate/heptanes (2×30 mL). The combined organic layers were dried over magnesium sulfate, filtered and the filtrate was concentrated. The residue was purified using flash chromatography (40 g silica column, 0-40% ethyl acetate/heptanes) to afford the title compound. 1H NMR (600 MHz, CDCl 3) δ ppm 7.40-7.26 (m, 5H), 4.59 (d, J=12.3 Hz, 1H), 4.54 (d, J=12.3 Hz, 1H), 4.15 (ddd, J=10.8, 4.7, 2.3 Hz, 1H), 3.52-3.39 (m, 3H), 3.36 (s, 3H), 3.32-3.23 (m, 1H), 3.13 (t, J=10.4 Hz, 1H), 2.24-2.16 (m, 1H), 1.76-1.68 (m, 1H), 1.46-1.30 (m, 2H). LC/MS (APCI+) m/z 237.46 (M+H) +.
A solution of Example 25D (0.97 g) in tetrahydrofuran (10.00 mL) was added to a flask containing Pd(OH) 2/C (20 weight % Pd, 50% moisture, 50 mg). The flask was purged with nitrogen and a hydrogen balloon was connected. The reaction was stirred at ambient temperature for 24 hours and then the mixture was filtered and concentrated. The residue was dissolved in dichloromethane (15 mL), then N,N-dimethylpyridin-4-amine (0.750 g), N-ethyl-N-isopropylpropan-2-amine (1.500 mL) and 4-methylbenzene-1-sulfonyl chloride (0.800 g) were added sequentially. The reaction was stirred at ambient temperature for 3 hours and then the mixture was poured over water (20 mL) and extracted with dichloromethane (2-20 mL). The combined organic layers were dried over magnesium sulfate, filtered and the filtrate was concentrated. The residue was purified using flash chromatography (silica, 0-100% ethyl acetate/heptanes) to afford the title compound. 1H NMR (500 MHz, CDCl 3) δ ppm 7.79 (d, J=8.5 Hz, 2H), 7.34 (d, J=7.9 Hz, 2H), 4.04 (ddd, J=10.8, 4.7, 2.3 Hz, 1H), 3.97 (d, J=5.1 Hz, 2H), 3.48 (dtd, J=10.7, 5.1, 2.5 Hz, 1H), 3.34 (s, 3H), 3.25-3.15 (m, 1H), 3.04 (dd, J=10.8, 10.1 Hz, 1H), 2.45 (s, 3H), 2.23-2.15 (m, 1H), 1.74-1.65 (m, 1H), 1.41-1.23 (m, 2H). LC/MS (APCI+) m/z 301.36 (M+H) +.
A mixture of Example 25E (1.000 g), N,N-dimethylformamide (6.00 mL) and sodium azide (1.000 g) was heated at 80° C. for 18 hours. The reaction was cooled to ambient temperature and poured over water (30 mL) and extracted with 5:1 ethyl acetate/heptanes (2×20 mL). The combined organic layers were dried over magnesium sulfate, filtered and the filtrate was concentrated to afford the title compound. 1H NMR (600 MHz, CDCl 3) δ ppm 4.13 (ddd, J=10.8, 4.7, 2.3 Hz, 1H), 3.44 (dddd, J=10.9, 6.6, 3.9, 2.2 Hz, 1H), 3.36 (s, 3H), 3.30-3.19 (m, 3H), 3.11 (t, J=10.5 Hz, 1H), 2.25-2.17 (m, 1H), 1.73-1.66 (m, 1H), 1.47-1.30 (m, 2H). LC/MS (APCI+) m/z 144.27 (M-N 2+H) +.
To a solution of 5,5-dimethylcyclohexane-1,3-dione (15 g) and formaldehyde (19.92 g) in dichloromethane (600 mL) was added boron trifluoride diethyl etherate (40.7 mL) over 10 minutes, and the reaction mixture was stirred at ambient temperature for 2.5 hours. The reaction mixture was quenched with the addition of saturated aqueous NaHCO 3 solution, the organic layer was separated, and the aqueous layer was extracted with additional dichloromethane. The organic layers were combined, washed with brine, dried over magnesium sulfate, filtered, and concentrated. The residue was taken up in heptanes/ethyl acetate (5:1), concentrated, treated with heptanes (300 mL), and filtered. The residue was chromatographed over silica gel (ISCO Gold®), eluting with a gradient of 0 to 16% ethyl acetate/heptanes to afford the title compound. 1H NMR (400 MHz, CDCl 3) δ ppm 5.13 (s, 2H), 4.43 (t, 2H), 2.28 (t, 2H), 2.22 (s, 2H), 1.08 (s, 6H). MS (DCI+) m/z 183.1 (M+H) +.
To a solution of 1-bromo-4-chlorobenzene (30.3 g) in tetrahydrofuran (200 mL) at −78° C. was added ii-butyllithium (2.5M in hexane, 60.6 mL) dropwise, keeping the temperature below −70° C. After stirring at −78° C. for 30 minutes, a solution of Example 25H (24 g) in tetrahydrofuran (75 mL) was added dropwise, keeping the temperature below −60° C. The reaction mixture was stirred at −78° C. for one hour, allowed to warm to ambient temperature and stirred for 12 hours. The reaction mixture was treated with 3 M aqueous HCl (80 mL) and stirred for 2 hours. Most of the organic solvent was removed and the resulting aqueous layer was extracted with ethyl acetate (three times). The combined organics were washed with brine, dried over magnesium sulfate, filtered, and concentrated. The residue was chromatographed over silica gel (750 g BIOTAGE® SNAP), eluting with a gradient of 5 to 22% ethyl acetate/heptanes to afford the title compound. 1H NMR (500 MHz, CDCl 3) δ ppm 7.43-7.32 (m, 2H), 7.28-7.21 (m, 2H), 4.20 (d, 2H), 2.85 (t, 1H), 2.57 (s, 2H), 2.42 (s, 2H), 1.14 (s, 6H). MS (ESI+) m/z 247.2 (M+H) +.
To a solution of Example 251 (15 g) in dichloromethane (800 mL) was added tetraethylammonium chloride (14.08 g) followed by triethylamine (11.85 mL). The reaction mixture was cooled to 0° C. with an ice/water bath and methane sulfonyl chloride (6.62 mL) was added over 10 minutes. The reaction mixture was allowed to warm to ambient temperature overnight. The reaction mixture was washed with saturated aqueous ammonium chloride solution and brine, dried over magnesium sulfate, filtered and concentrated. The residue was chromatographed over silica gel (330 g ISCO Gold®), eluting with a gradient of 0 to 35% ethyl acetate/heptanes to afford the title compound. 1H NMR (500 MHz, dimethyl sulfoxide-d 6) δ ppm 7.60-7.55 (m, 2H), 7.45-7.40 (m, 2H), 4.12 (s, 2H), 2.64 (s, 2H), 2.39 (s, 2H), 1.05 (s, 6H). MS (DCI+) m/z 283.1 (M+H) +.
To a solution of (S)-2-methyl-CBS-oxazaborolidine (20.96 g) in anhydrous tetrahydrofuran (230 mL) at −50° C. was added BH 3-tetrahydrofuran (1.0 M in tetrahydrofuran, 76 mL) over 45 minutes, and the solution was stirred for an additional 40 minutes. A solution of Example 25J (21 g) in anhydrous tetrahydrofuran (230 mL) was added dropwise via an addition funnel over the course of about 60 minutes. After the addition, the reaction mixture was stirred 1.5 hours at −50° C. The reaction mixture was quenched by the dropwise addition of methanol (130 mL) over about 20 minutes at −50° C. The cooling bath was removed, and the reaction mixture was allowed to warm to ambient temperature overnight. The reaction mixture was diluted with saturated aqueous NH 4Cl solution (250 mL), diluted with water and extracted with ethyl acetate. The organic layer was washed with additional aqueous saturated NH 4Cl and brine, dried over sodium sulfate, filtered, and concentrated. The residue was chromatographed over silica gel (Teledyne Isco RediSep® Rf GOLD® 330 g), eluting with a gradient of 0 to 20% ethyl acetate/heptanes to afford the title compound. 1H NMR (400 MHz, dimethyl sulfoxide-d 6) δ ppm 7.44-7.36 (m, 2H), 7.21-7.13 (m, 2H), 4.40-4.28 (m, 2H), 3.85 (d, 1H), 2.24 (dt, 1H), 1.82 (d, 1H), 1.74 (ddd, 1H), 1.43 (dd, 1H), 0.94 (s, 3H), 0.89 (s, 3H). MS (DCI+) m/z 284.1 (M+H) +.
To a solution of Example 25L (100 g) in dichloroethane (2150 mL) was added propane-1,3-diyl bis(trifluoromethanesulfonate) (95 g) followed by N 1,N 1,N 8,N 8-tetramethylnaphthalene-1,8-diamine (122 g) (proton sponge) and the reaction mixture was heated to 50° C. for 20 hours. The reaction mixture was cooled in an ice bath and filtered. The filtrate was washed with 1N aqueous HCl (1 L×3), 1N aqueous NaOH (three times), dried over magnesium sulfate, filtered and concentrated. The residue was chromatographed over silica gel (3 kg) with a gradient of 2 L of 5% ethyl acetate/heptanes, 4 L 10% ethyl acetate/heptanes, then 6-8 L 20% ethyl acetate/heptanes to afford the title compound. 1H NMR (400 MHz, dimethyl sulfoxide-d 6) δ ppm 7.38-7.30 (m, 2H), 7.09-7.01 (m, 2H), 4.03-3.93 (m, 1H), 3.83-3.57 (m, 7H), 3.46-3.36 (m, 2H), 2.66 (d, 2H), 2.19 (d, 1H), 2.06 (d, 1H), 1.92-1.72 (m, 4H), 1.65-1.55 (m, 2H), 1.49-1.33 (m, 2H), 1.32 (s, 9H), 0.94 (s, 3H), 0.94 (s, 3H). MS (ESI+) m/z 505.3 (M+H) +.
To a solution of Example 25M (90 g) in dichloromethane (713 mL) cooled in an ice bath was added trifluoroacetic acid (206 mL) dropwise over about 20 minutes. The ice bath was removed, and the reaction mixture was stirred for 3 hours. The reaction mixture was concentrated, and the residue was dissolved in ethyl acetate (300 mL). The vigorously stirred solution was neutralized by the addition of saturated aqueous Na 2CO 3 (350 mL). The organic layer was separated, washed four times with saturated aqueous Na 2CO 3, dried over sodium sulfate, filtered, and concentrated to afford the title compound. 1H NMR (400 MHz, dimethyl sulfoxide-d 6) δ 7.45-7.33 (m, 2H), 7.13-7.02 (m, 2H), 3.87 (q, J=8.0 Hz, 1H), 3.79-3.62 (m, 4H), 3.48-3.36 (m, 2H), 2.79-2.53 (m, 4H), 2.19 (d, J=12.1 Hz, 1H), 2.09 (dt, J=17.1, 3.0 Hz, 1H), 1.95, 1.87 (m, 2H), 1.85 (dd, J=6.5, 1.8 Hz, 1H), 1.78-1.70 (m, 1H), 1.63 (dq, J=7.5, 3.9 Hz, 2H), 1.47 (td, J=11.3, 2.8 Hz, 1H), 1.39 (dd, J=12.5, 9.1 Hz, 1H), 0.98 (s, 4H), 0.97 (s, 3H). MS (DCI+) m/z 405.3 (M+H) +.
A 2 L three-neck round bottom flask equipped with a stir bar, heating mantle, nitrogen inlet and outlet, and a thermocouple was charged with ethyl 2-bromo-4-fluorobenzoate (44.4 g). Anhydrous dimethyl sulfoxide (346 mL) was added to the flask, and mixture stirred at ambient temperature. Example 25N (70 g) was added, followed by potassium hydrogenphosphate (90 g). Once the slurry was mixed thoroughly, the temperature was increased to 60° C., and the slurry was heated under nitrogen for 72 hours. The heating mantle was removed, and the reaction was cooled with an ice/water bath to 8° C. The flask was equipped with a 1 L addition funnel. To this cold reaction slurry was added water (850 mL) dropwise via an addition funnel, and the mixture was sonicated for 30 minutes. The mixture was subjected to mechanical stirring and stirred vigorously for 1 hour at ambient temperature. The precipitate was filtered through a Buchner funnel loaded with filter paper. The filtered solids were washed with water (2×500 mL) and allowed to dry on the filter for 16 hours. The solids were dissolved in ethyl acetate (600 mL), and water (300 mL) was added to the solution. The two-phase solution was stirred for 1 hour. The layers were separated in a separatory funnel, and organic layer washed with water (300 mL) and brine (200 mL). The organic layer was dried with 100 g of magnesium sulfate, filtered and concentrated to afford the crude residue. The residue was dissolved in dichloromethane (100 mL) and purified via normal phase chromatography using a BIOTAGE® Snap Ultra 750 g. silica gel column eluting with a gradient of 0 to 30% ethyl acetate in heptane to afford the title compound. 1H NMR (400 MHz, dimethyl sulfoxide-d 6) δ ppm 7.68 (d, J=8.9 Hz, 1H), 7.46-7.33 (m, 2H), 7.11 (d, J=2.6 Hz, 1H), 7.10-7.07 (m, 2H), 6.91 (dd, J=9.0, 2.5 Hz, 1H), 4.22 (q, J=7.1 Hz, 2H), 4.04 (t, J=7.7 Hz, 1H), 3.89-3.64 (m, 7H), 3.59 (d, J=12.0 Hz, 1H), 3.48 (q, J=8.3 Hz, 1H), 2.95 (dd, J=12.2, 10.8 Hz, 1H), 2.86-2.66 (m, 2H), 2.27 (d, J=12.2 Hz, 1H), 2.15-2.06 (m, 1H), 2.01 (d, J=10.6 Hz, 1H), 1.97-1.83 (m, 2H), 1.75-1.57 (m, 3H), 1.40 (dd, J=12.5, 9.1 Hz, 1H), 1.28 (t, J=7.1 Hz, 3H), 0.99 (d, J=1.9 Hz, 6H). LC/MS (APCI+) m/z 631.46 (M+H) +.
Example 25P
5-bromo-1H-pyrrolo[2,3-b]pyridine 7-oxide
To a solution of 5-bromo-1H-pyrrolo[2,3-b]pyridine (10 g) in ethyl acetate (200 mL) was added 3-chloroperoxybenzoic acid (21.90 g) at 25° C., then stirred at 25° C. for 3 hours. The reaction mixture was diluted with ethyl acetate (100 mL), then quenched by addition of saturated sodium bicarbonate (1000 mL). The biphasic mixture was filtered, and the filter cake was washed with water (100 mL) and then dried in vacuo to afford the title compound. 1H NMR (400 MHz, dimethyl sulfoxide-d 6) δ ppm 8.32 (s, 1H), 8.41-8.22 (m, 1H), 7.86 (s, 1H), 7.47 (s, 1H), 6.50 (d, J=1.8 Hz, 1H).
Example 25Q
5-bromo-6-chloro-1H-pyrrolo[2,3-b]pyridine
To a solution of Example 25P (30 g) and 1,1,1,3,3,3-hexamethyldisilazane (29.5 mL) in tetrahydrofuran (300 mL) was added 2,2,2-trichloroacetyl chloride (47.1 mL) at 0° C. The resulting mixture was stirred for 0.5 hours and warmed to 25° C. for another 2 hours. The reaction mixture was concentrated under reduced pressure. The residue was diluted with water (200 mL) and extracted with ethyl acetate (3×100 mL). The combined organic layers were washed with brine (100 mL), dried over sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to afford a residue which was triturated with ethyl acetate and petroleum ether (1:10, 100 mL) to afford the title compound. 1H NMR (400 MHz, dimethyl sulfoxide-d 6) δ ppm 12.05 (s, 1H), 8.41 (s, 1H), 7.63-7.54 (m, 1H), 6.51-6.45 (m, 1H).
To a solution of Example 25Q (15 g) in N,N-dimethylformamide (150 mL) was added sodium hydride (3.11 g) in portions at 0° C. The resulting mixture was stirred at 0° C. for 1 hour, then a solution of 2-(trimethylsilyl)ethoxymethyl chloride (13.79 mL) in N,N-dimethylformamide (50 mL) was added dropwise. The resulting mixture was stirred at 0° C. for another 2 hours. The reaction mixture was diluted with brine (200 mL) and extracted with ethyl acetate (3×100 mL). The combined organic layers were washed with brine (100 mL), dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to afford a residue, which was purified by column chromatography on silica gel (eluted with petroleum ether:ethyl acetate=10:1) to afford the title compound. 1H NMR (400 MHz, CDCl 3) δ ppm 8.15 (s, 1H), 7.36 (d, J=2.4 Hz, 1H), 6.48 (d, J=2.1 Hz, 1H), 5.61 (s, 2H), 3.54 (t, J=7.9 Hz, 2H), 0.92 (t, J=7.9 Hz, 2H), 0.01 (s, 9H).
To a solution of N-(3-hydroxypropyl)-4-methylbenzenesulfonamide (0.349 g) in anhydrous tetrahydrofuran (5 mL) was added sodium hydride (0.166 g) in portions at 0° C. The resulting mixture was stirred at 0° C. for 0.5 hours, then Example 25R (0.5 g) was added. The reaction mixture was heated to 80° C. and stirred for 12 hours under nitrogen. After cooling, the reaction was diluted with water (100 mL), then extracted with ethyl acetate (3×100 mL). The combined organic layers were washed with brine (100 mL), dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to afford a residue, which was purified by column chromatography on silica gel (eluted with petroleum ether:ethyl acetate=5:1) to afford the title compound. 1H NMR (400 MHz, CDCl 3) δ ppm 8.03 (s, 1H), 7.76 (d, J=8.2 Hz, 2H), 7.23 (d, J=8.1 Hz, 2H), 7.15 (d, J=3.5 Hz, 1H), 6.42-6.37 (m, 1H), 5.53 (s, 2H), 4.47-4.41 (m, 2H), 3.55-3.48 (m, 2H), 3.23 (q, J=6.0 Hz, 2H), 2.37 (s, 3H), 2.01 (J=5.7 Hz, 2H), 0.90-0.87 (m, 2H), ), −0.03 (s, 9H).
To a solution of Example 25S (500 mg) in dimethyl sulfoxide (6 mL) was added potassium carbonate (374 mg), picolinic acid (89 mg) and copper(I) iodide (206 mg) at 20° C. The reaction mixture was stirred at 160° C. under microwave for 2 hours. After cooling to ambient temperature, the reaction was diluted with water (100 mL), then extracted with ethyl acetate (3×50 mL). The combined organic layers were washed with brine (50 mL), dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to afford a residue, which was purified by column chromatography on silica gel (eluted with petroleum ether:ethyl acetate=3:1) to afford the title compound. 1H NMR (400 MHz, CDCl 3) δ ppm 8.13 (s, 1H), 7.48 (d, J=8.1 Hz, 2H), 7.31 (d, J=3.5 Hz, 1H), 7.20 (d, J=8.1 Hz, 2H), 6.52 (d, J=3.5 Hz, 1H), 5.58 (s, 2H), 4.03-3.78 (m, 4H), 3.60-3.44 (m, 2H), 2.39 (s, 3H), 1.90 (s, 2H), 0.90 (t, J=8.2 Hz, 2H), −0.05 (s, 9H).
To a solution of sodium (0.291 g) in 1,2-dimethoxyethane (0.5 mL) was added naphthalene (1.624 g) under nitrogen. The mixture was stirred at 20° C. for 1 hour until the formation of sodium/naphthalene was complete. Then to the solution of Example 25T (1 g) in anhydrous tetrahydrofuran (10 mL) was added to the above solution at −78° C. The resulting mixture was brought to 20° C. and stirred for 2 hours. The reaction was quenched by addition of water (200 mL) and extracted with ethyl acetate (3×100 mL). The combined organic layers were washed with brine (100 mL), dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to afford a residue, which was purified by preparative-HPLC to afford the title compound. 1H NMR (400 MHz, CDCl 3) δ ppm 8.17 (s, 1H), 7.80 (s, 3H), 7.37 (d, J=3.5 Hz, 1H), 6.51 (d, J=3.5 Hz, 1H), 5.59 (s, 2H), 4.38-4.27 (m, 2H), 3.61-3.46 (m, 4H), 2.41 (s, 2H), 0.93-0.86 (m, 2H), −0.06 (s, 9H).
To an oven dried 2 L three-necked round bottom flask equipped with a mechanical stirrer, a Huber-chilled reflux condenser, Claisen head adapter, nitrogen needle inlet and outlet to bubbler through a septa, and thermocouple was charged Example 25U (79 g), (Example 1N) (26.6 g), and methanesulfonato[9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene](2′-methylamino-1,1′-biphenyl-2-yl)palladium(II) (5.61 g). These solids were put under a heavy flow of nitrogen, and then cesium carbonate (81 g) was added quickly to the reaction flask. The solids were mixed slowly with the mechanical stirrer, and the heavy flow of nitrogen through the reaction flask was continued for 60 minutes. In a separate oven dried 2 L round bottom flask equipped with a stir bar and septum was charged anhydrous toluene (833 mL). This solvent was sparged subsurface with a heavy nitrogen flow for 60 minutes while stirring. The solvent was then transferred via cannula to the three-necked flask, and the reaction was heated to an internal temperature of 110° C. under a flow of nitrogen for 16 hours. The reaction was cooled to ambient temperature, and the flask was charged with water (600 mL), followed by ammonium pyrrolidinedithiocarbamate palladium scavenger (3 g). This mixture was stirred vigorously for 1 hour. The reaction was diluted further with ethyl acetate (400 mL), stirred for 30 minutes, and then filtered through a plug of diatomaceous earth. The filter cake was washed with ethyl acetate (2×500 mL). The filtrate was transferred to a separatory funnel and the layers separated. The organic layer washed with water (200 mL), and then brine (200 mL). The combined aqueous layers were back extracted one time with ethyl acetate (200 mL). The combined organic layers were dried with sodium sulfate (200 g), filtered, and concentrated to produce the crude residue. The residue was dissolved in dichloromethane (200 mL) and purified via normal phase chromatography using a BIOTAGE® Snap Ultra 1.5 kg. silica gel column eluting with a gradient of 0 to 50% ethyl acetate in heptane to afford the title compound. 1H NMR (500 MHz, dimethyl sulfoxide-d 6) δ ppm 7.44 (d, J=8.8 Hz, 1H), 7.40-7.35 (m, 2H), 7.30 (d, J=3.5 Hz, 1H), 7.12-7.07 (m, 2H), 7.03 (s, 1H), 6.63 (d, J=2.4 Hz, 1H), 6.58 (dd, J=8.9, 2.3 Hz, 1H), 6.22 (d, J=3.5 Hz, 1H), 5.43 (s, 2H), 4.47-4.37 (m, 2H), 4.03 (t, J=8.0 Hz, 1H), 3.89-3.69 (m, 7H), 3.66 (q, J=5.7, 4.5 Hz, 3H), 3.59 (t, J=11.1 Hz, 2H), 3.52-3.42 (m, 3H), 2.96-2.87 (m, 1H), 2.85-2.77 (m, 1H), 2.67 (td, J=11.9, 3.0 Hz, 1H), 2.27 (d, J=12.1 Hz, 1H), 2.11 (dt, J=17.4, 3.0 Hz, 1H), 2.02 (d, J=10.8 Hz, 1H), 2.00-1.85 (m, 4H), 1.71 (td, J=11.6, 2.9 Hz, 1H), 1.64 (dd, J=8.7, 4.8 Hz, 2H), 1.40 (dd, J=12.4, 9.1 Hz, 1H), 0.98 (s, 6H), 0.94 (t, J=7.1 Hz, 3H), 0.84-0.79 (m, 2H), −0.08 (s, 9H).
To a 2 L three-necked flask equipped with a mechanical stirrer, heating mantle, Claisen head adapter, reflux condenser, nitrogen inlet and outlet to bubbler, and a thermocouple was charged Example 25V (69 g). The solids were dissolved in anhydrous tetrahydrofuran (330 mL). To this solution at ambient temperature was added ethylenediamine (53.5 mL) and tetrabutyl ammonium fluoride (1.0 M in tetrahydrofuran, 793 mL). The reaction was heated to 66° C. internal temperature for 24 hours. The heating mantle was removed, and the reaction cooled to 8° C. in an ice/water bath. The mixture was quenched with water (200 mL). The reaction mixture was diluted with ethyl acetate (200 mL) and then partitioned in a separatory funnel. The organic layer was washed with water (200 mL) and brine (200 mL). The organic layer was dried with sodium sulfate (100 g), filtered and concentrated in vacuo to afford the crude product. The residue was suspended in 1:1 methyl tert-butyl ether:heptane (400 mL), sonicated for 30 minutes then stirred vigorously for 1 hour. The solids were filtered, and the filter cake washed with 1:1 methyl tert-butyl ether heptane (50 mL). The solids were dried on the Buchner funnel to afford the title compound. 1H NMR (600 MHz, dimethyl sulfoxide-d 6) δ ppm 11.13 (t, J=2.2 Hz, 1H), 7.41 (d, J=8.8 Hz, 1H), 7.40-7.37 (m, 2H), 7.14 (dd, J=3.4, 2.4 Hz, 1H), 7.11-7.07 (m, 2H), 7.02 (d, J=0.7 Hz, 1H), 6.60 (d, J=2.4 Hz, 1H), 6.55 (dd, J=8.9, 2.3 Hz, 1H), 6.13 (dd, J=3.4, 1.9 Hz, 1H), 4.44-4.33 (m, 2H), 4.03 (tt, J=7.1, 3.5 Hz, 1H), 3.90-3.69 (m, 4H), 3.68-3.62 (m, 3H), 3.61-3.54 (m, 2H), 3.51-3.44 (m, 1H), 2.92-2.86 (m, 1H), 2.84-2.78 (m, 1H), 2.66 (td, J=11.9, 3.0 Hz, 1H), 2.32-2.28 (m, 1H), 2.26 (d, J=12.1 Hz, 1H), 2.11 (d, J=17.1 Hz, 1H), 2.02 (d, J=10.8 Hz, 1H), 1.98-1.86 (m, 4H), 1.70 (td, J=11.7, 3.0 Hz, 1H), 1.66-1.58 (m, 2H), 1.40 (dd, J=12.5, 9.2 Hz, 1H), 1.36-1.30 (m, 1H), 1.29-1.22 (m, 1H), 0.98 (s, 6H), 0.95 (t, J=7.1 Hz, 3H).
To a 5 L three-necked flask equipped with a mechanical stirrer, heating mantle, reflux condenser, Claisen adapter, nitrogen inlet and outlet to bubbler, and a thermocouple was charged Example 25W (58.7 g). The residue was dissolved in 1,4-dioxane (991 mL) and methanol (496 mL) and stirred at ambient temperature for 5 minutes. Then lithium hydroxide (18.99 g) and water (496 mL) were then added, and the reaction was heated to an internal temperature of 75° C. for 16 hours. The heating mantle was removed, and the reaction was cooled to 5° C. in an ice/water bath. The reaction was neutralized to pH 7 by careful addition of 3N aqueous hydrochloric acid (100 mL). The pH was adjusted further to pH 6 with the addition of saturated aqueous ammonium chloride (100 mL). The mixture was diluted with dichloromethane (300 mL) and the layers partitioned in a separatory funnel. The aqueous layer was extracted with dichloromethane (2×100 mL), and the organic layers combined and dried with sodium sulfate (50 g). The solids were filtered, and the filtrate concentrated in vacuo to produce the crude residue. The residue was dissolved in dichloromethane (50 mL) and purified via normal phase chromatography on a silica gel cartridge (Teledyne Isco RediSep® RF GOLD®, 330 g) eluting with 0 to 10% methanol in dichloromethane to afford the title compound. 1H NMR (500 MHz, dimethyl sulfoxide-d 6) δ ppm 12.28 (s, 1H), 11.20 (t, J=2.2 Hz, 1H), 7.62 (d, J=8.8 Hz, 1H), 7.43-7.35 (m, 2H), 7.17 (dd, J=3.4, 2.5 Hz, 1H), 7.12-7.06 (m, 2H), 7.01 (d, J=0.7 Hz, 1H), 6.71 (d, J=2.4 Hz, 1H), 6.68 (dd, J=8.9, 2.4 Hz, 1H), 6.15 (dd, J=3.3, 1.9 Hz, 1H), 4.34 (t, J=5.5 Hz, 2H), 4.03 (t, J=7.7 Hz, 1H), 3.88-3.75 (m, 3H), 3.74-3.64 (m, 3H), 3.64-3.58 (m, 1H), 3.57 (s, 6H), 3.51-3.43 (m, 1H), 2.92 (t, J=11.4 Hz, 1H), 2.85-2.78 (m, 1H), 2.68 (td, J=12.0, 3.0 Hz, 1H), 2.26 (d, J=12.1 Hz, 1H), 2.11 (d, J=17.4 Hz, 1H), 2.02 (d, J=10.9 Hz, 1H), 1.97 (p, J=5.8 Hz, 1H), 1.95-1.85 (m, 2H), 1.70 (td, J=11.6, 2.9 Hz, 1H), 1.66-1.60 (m, 0H), 1.40 (dd, J=12.5, 9.1 Hz, 1H), 0.98 (s, 6H). LC/MS (APCI+) m/z 712.29 (M+H) +.