It's only fair to share...














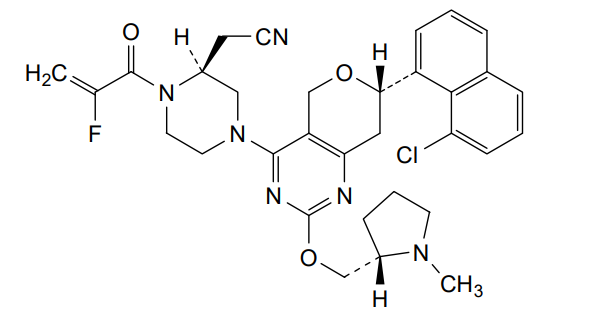
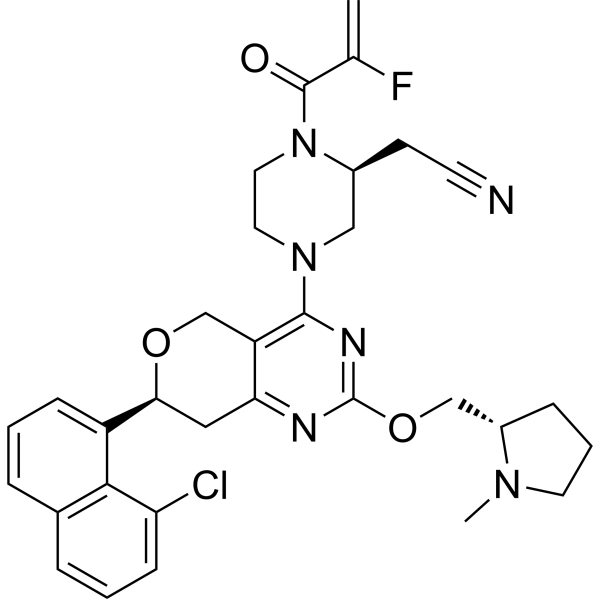

Talorasib
CAS 2648584-48-7
MFC32H34ClFN6O3 MW605.10
- (2S)-4-[(7S)-7-(8-Chloro-1-naphthalenyl)-7,8-dihydro-2-[[(2S)-1-methyl-2-pyrrolidinyl]methoxy]-5H-pyrano[4,3-d]pyrimidin-4-yl]-1-(2-fluoro-1-oxo-2-propen-1-yl)-2-piperazineacetonitrile
- [(2S)-4-[(7S)-7-(8-chloronaphthalen-1-yl)-2-{[(2S)-1-methylpyrrolidin-2-yl]methoxy}-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-4-yl]-1-(2-fluoroprop-2-enoyl)piperazin-2-yl]acetonitrile
- 2-Piperazineacetonitrile, 4-[(7S)-7-(8-chloro-1-naphthalenyl)-7,8-dihydro-2-[[(2S)-1-methyl-2-pyrrolidinyl]methoxy]-5H-pyrano[4,3-d]pyrimidin-4-yl]-1-(2-fluoro-1-oxo-2-propen-1-yl)-, (2S)-
[(2S)-4-[(7S)-7-(8-chloronaphthalen-1-yl)-2-{[(2S)-1-methylpyrrolidin-2-yl]methoxy}-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-4-yl]-1-(2-fluoroprop-2-enoyl)piperazin-2-yl]acetonitrile
Kirsten rat sarcoma viral oncogene homologue (KRAS)inhibitor, antineoplastic, 727W6T7DPK
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN380619664&_cid=P20-MJ0TAW-52678-1
| Preparation Example 1: Synthesis of the compound shown in formula (I) |
| (1) Synthesis of Compound 1 |
| Synthetic route of compound 1: |
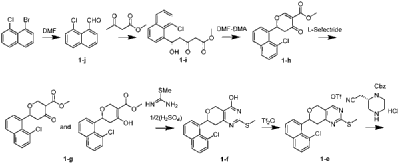
| Synthesis of compound 1-j |
| 1-Bromo-8-chloronaphthalene (500 mg, 2.07 mmol) was dissolved in THF (20 mL), cooled to -78 °C, and n-BuLi (2.5 M, 1.66 mL, 4.14 mmol) was added dropwise under nitrogen protection. After the addition was complete, the mixture was stirred at -78 °C for 10 min, and then DMF (800 μL, 10.35 mmol) was added dropwise at -78 °C. After the addition was complete, the reaction mixture was stirred at -78 °C for 30 min, then heated to room temperature and stirred for 2 h. The reaction was quenched with 50 mL of saturated ammonium chloride solution and extracted with ethyl acetate (50 mL * 2). The organic phase was washed with saturated brine (50 mL * 2), treated with anhydrous sodium sulfate, filtered, and concentrated to obtain the crude product. The crude product was purified by rapid column chromatography (EA/PE = 1/10) to give compound 1-j (330 mg, 84% yield) as a white solid. LC-MS (ESI): m/z=191.0[M+H] + ; 1 H NMR (400MHz, CDCL 3 ):δ11.31(s,1H),8.03(dd,1H,J 1 =1.2Hz,J 2 =8.4Hz), 7.92(dd,1H,J 1 =1.2Hz,J 2 =7.2Hz),7.86(1H,J=8.4Hz),7.70(dd,1H,J 1 =1.2Hz,J 2 =7.6Hz), 7.59(t,1H,J=7.6Hz), 7.47(t,1H,J=8Hz). |
| Synthesis of compound 1-i |
| At room temperature, NaH (60%, 242 mg, 6.05 mmol) was added to 6 mL of THF. Then, methyl acetoacetate (543 μL, 5.04 mmol) was added under nitrogen atmosphere at room temperature. The mixture was stirred for 30 minutes under nitrogen atmosphere at room temperature, and then n-BuLi (2.5 M, 2.4 mL, 6.05 mmol) was added dropwise at -15 °C to -10 °C. After the addition was complete, the mixture was maintained at this temperature for 30 minutes, and then a 10 mL solution of compound 1-j (320 mg, 1.68 mmol) in THF was added dropwise. After the addition was complete, the mixture was stirred at low temperature (-10 °C to 0 °C) for 2 hours, then quenched with saturated ammonium chloride solution (50 mL), and then extracted with ethyl acetate (50 mL x 2). The organic phase was washed with saturated brine (50 mL * 2), treated with anhydrous sodium sulfate, filtered, and concentrated to obtain the crude product. The crude product was purified by rapid column chromatography (EA/DCM = 1/10) to give compound 1-i (510 mg, 99% yield) as a white solid. LC-MS (ESI): m/z = 329.1 [M + Na] ⁺ ; 1H NMR (400 MHz, CDCl₂) 3 ): δ8.06(d,1H,J=6.4Hz),7.79(d,2H,J=8Hz),7.58(dd,1H,J 1 =7.6Hz,J 2 =1.6Hz),7.53(t,1H,J=7.6Hz),7.34(t,1H,J=7.6Hz),6.91(dd,1H,J 1 =9.2Hz, J 2 =2.4Hz),3.74(s,3H),3.54(s,2H),3.36(dd,1H,J 1 =18Hz,J 2 =1.6Hz),3.24(d,1H,J=3.6Hz),2.85-2.75(m,1H). |
| Synthesis of compound 1-h |
| Compound 1-i (510 mg, 1.66 mmol) was dissolved in DCM (18 mL) at room temperature, followed by the addition of DMF-DMA (245 μL, 1.83 mmol) under nitrogen atmosphere at room temperature. After stirring the reaction mixture for 45 minutes at room temperature, BF was added. 3 Et 2 O (232 μL, 1.83 mmol). After addition, the mixture was stirred at room temperature for 1 hour, then diluted with 100 mL of ethyl acetate. The organic phase was then sequentially quenched with saturated NaHCO3. 3 The sample was washed with a solution (100 mL) and saturated saline solution (100 mL * 2), treated with anhydrous sodium sulfate, filtered, and concentrated to obtain the crude compound 1-h (520 mg). The crude product required no purification and was used directly in the next reaction. LC-MS (ESI): m/z = 317.1 [M+1] + . |
| Synthesis of compound 1-g |
| Compound 1-h (520 mg, 1.64 mmol) was dissolved in THF (20 mL) at room temperature, and then tri-sec-butylborohydride (1 M, 1.64 mL, 1.64 mmol) was added dropwise under nitrogen atmosphere at -78 °C. After addition, the mixture was stirred at -78 °C for 1 hour, the reaction was quenched with saturated ammonium chloride solution (50 mL), extracted with ethyl acetate (50 mL * 2), the organic matter was washed with saturated brine (50 mL * 2), treated with anhydrous sodium sulfate, filtered, and concentrated to obtain the crude product. The crude product was purified by rapid column chromatography (PE/EA = 4/1) to give compound 1-g (338 mg, 65% yield) as a yellow oil. LC-MS (ESI): m/z = 319.0 [M+1] + . |
| Synthesis of compound 1-f |
| Compound 1-g (338 mg, 1.06 mmol) was dissolved in methanol (20 mL) at room temperature. Then, under nitrogen atmosphere at 0 °C, sodium methoxide (286 mg, 5.3 mmol) and compound 2-methyl-2-mercaptourea sulfate (265 mg, 0.954 mmol) were added sequentially. After the addition was complete, the mixture was brought to room temperature and stirred for 20 hours. The pH of the reaction solution was adjusted to 5 with 1 N dilute hydrochloric acid, and a solid precipitated. The solid was filtered, the filter cake was washed with water (5 mL * 2), and the solid was collected and dried under vacuum to give crude product 1-f (313 mg) as a white solid. LC-MS (ESI): m/z = 359.1 [M+1] + . |
| Synthesis of compound 1-e |
| Compound 1-f (313 mg, 0.87 mmol) was dissolved in DCM (10 mL) at room temperature. Then, under nitrogen atmosphere in an ice-water bath, DIPEA (431 μL, 2.61 mmol) and trifluoromethanesulfonic anhydride (219 μL, 1.31 mmol) were added sequentially. After addition, the reaction mixture was stirred in an ice-water bath for 2 hours, quenched with saturated sodium bicarbonate solution (50 mL), extracted with DCM (50 mL x 2), and the organic phase was treated with anhydrous sodium sulfate, filtered, and concentrated to obtain a crude product. The crude product was purified by rapid column chromatography (EA/PE = 1/10) to give compound 1-e (83 mg, 16% yield in 2 steps) as a white solid. LC-MS (ESI): m/z = 491.0 [M+1] + . |
| Synthesis of compound 1-d |
| Compound 1-e (83 mg, 0.169 mmol) was dissolved in DMF (10 mL) at room temperature, followed by the sequential addition of DIPEA (84 μL, 0.507 mmol) and (S)-2-cyanomethylpiperazine-1-carboxylate hydrochloride (59.9 mg, 0.203 mmol). After addition, the mixture was stirred for 1 hour at 100 °C under nitrogen protection, cooled to room temperature, quenched with saturated brine (50 mL), and extracted with ethyl acetate (50 mL x 2). The organic phase was washed with saturated brine (50 mL x 3), treated with anhydrous sodium sulfate, filtered, and concentrated to obtain a crude product. The crude product was purified by rapid column chromatography (EA/PE = 1/1) to give compound 1-d (101 mg, 99% yield) as a white solid. LC-MS (ESI): m/z = 600.2 [M+1] + . |
| Synthesis of compound 1-c |
| Compound 1-d (101 mg, 0.168 mmol) was dissolved in ethyl acetate (10 mL) at room temperature, followed by the addition of MCPBA (85%, 88.4 mg, 0.437 mmol) at room temperature. After addition, the mixture was stirred at room temperature for 2 hours, quenched with saturated sodium bicarbonate solution (20 mL), extracted with ethyl acetate (25 mL x 2), and the organic phase was treated with anhydrous sodium sulfate, filtered, and concentrated to obtain a crude product. The crude product was purified by rapid column chromatography (EA/PE = 1/4) to give compound 1-c (88 mg, 82% yield) as a white solid. LC-MS (ESI): m/z = 632.1 [M+1] + . |
| Synthesis of compound 1-b |
| Compound 1-c (88 mg, 0.139 mmol) was dissolved in toluene (10 mL) at room temperature. The reaction mixture was then cooled to 0 °C, and N-methylprolyl (29 μL, 0.243 mmol) and t-BuONa (27 mg, 0.278 mmol) were added sequentially. After the addition was complete, the reaction mixture was stirred for 0.5 hours under nitrogen in an ice-water bath, quenched with water (20 mL), and extracted with ethyl acetate (30 mL * 2). The organic phase was treated with anhydrous sodium sulfate, filtered, and concentrated to obtain a crude product. The crude product was purified by rapid column chromatography (MeOH/DCM = 1/10) to give compound 1-b (78 mg, 84% yield) as a white solid. LC-MS (ESI): m/z = 667.3 [M+1] + . |
| Synthesis of compound 1-a |
| Compound 1-b (72 mg, 0.108 mmol) was dissolved in methanol (50 mL) at room temperature. The reaction solution was then cooled to -78 °C, purged twice with nitrogen, and then Pd/C (150 mg) and ZnBr were added. 2 (24.3 mg, 0.108 mmol), the reaction mixture was purged with hydrogen three times, brought to room temperature, and stirred under hydrogen atmosphere for 5 hours. The reaction mixture was filtered and concentrated to obtain a crude product, which was then purified by a rapid separation column (MeOH/DCM = 1:4) to give compound 1-a (20 mg, 35% yield) as a white solid. LC-MS (ESI): m/z = 533.0 [M+1] + . |
| Synthesis of Compound 1 |
| At room temperature, compound 2-fluoroacrylic acid (5.1 mg, 0.0563 mmol) was dissolved in DMF (2 mL). Then, at 0 °C, HATU (25.6 mg, 0.0675 mmol) and DIPEA (18.6 μL, 0.113 mmol) were added sequentially. After the addition was complete, the reaction mixture was stirred at 0 °C under nitrogen for 20 minutes. Then, a DMF solution of compound 1-a (20 mg, 0.0375 mmol) (3 mL) was added to the above reaction mixture. The mixture was brought to room temperature and stirred for another 5 hours. The reaction mixture was quenched with saturated brine (20 mL), extracted with ethyl acetate (25 mL * 2), washed with saturated brine (50 mL * 3), treated with anhydrous sodium sulfate, filtered, and concentrated to obtain the crude product. The crude product was purified by PREP-TLC (MeOH/DCM = 1/10) to obtain compound 1 (6 mg, 26% yield) as a white solid. LC-MS (ESI): m/z=605.2[M+1] + ; 1 H NMR (400MHz, CDCl 3 ): δ7.99-7.93(m,1H),7.83(t,2H,J=8.8Hz),7.62-7.49(m,2H),7.36(t,1H,J=7.6Hz),6.5 5-6.44(m,1H),5.51-5.31(m,1H),5.25(d,1H,J=16.8Hz),5.02-4.93(m,1H),4.82(dd,1H,J 1 =2.4Hz, J 2 =13.6Hz),4.48-4.38(m,1H),4.32-4.19(m,1H),4.17-4.04(m,1H),4. 00(d,1H,J=14Hz),3.87-3.70(m,1H),3.66-3.36(m,2H),3.31-3.16(m ,2H),3.14-2.98(m,1H),2.96-2.69(m,4H),2.59(d,3H,J=18Hz),2.52 -2.34(m,1H),2.15-2.06(m,1H),1.87-1.74(m,2H),0.93-0.76(m,2H). |
| (2) Resolution of compound 1 |
| Synthesis of compounds 1-1 and 1-2 |
| |
| The challenge lay in obtaining the compound shown in formula (I) through chiral resolution of compound 1. Despite trying various conditions, the two isomers of compound 1 could not be separated on a thin-layer chromatography plate, making separation impossible by thin-layer chromatography. Even in HPLC, the separation of the two isomers of compound 1 was poor, making separation impossible by preparative HPLC. Finally, chiral resolution had to be resorted to. After trying several conditions (as shown in Table 1 below), chiral resolution condition 9 was finally found, which enabled the separation of the compound shown in formula (I) and its diastereomers. |
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022081655&_cid=P20-MJ0T2K-48115-1
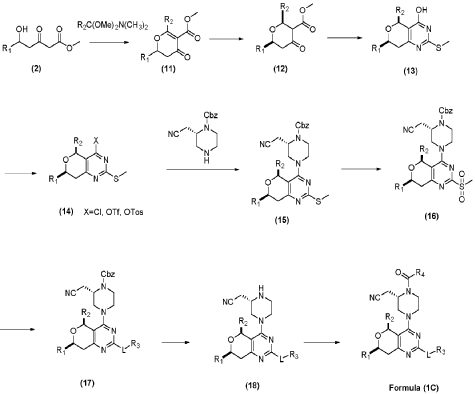
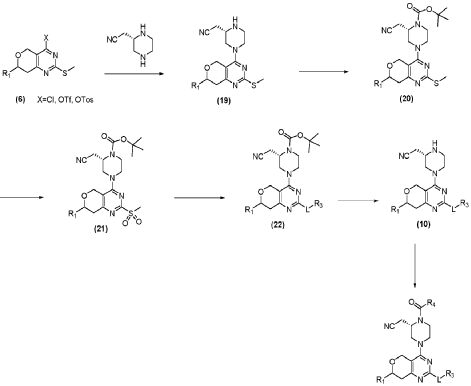
PAT
- Substituted dihydropyranopyrimidine compounds as kras inhibitorsPublication Number: US-2022112204-A1Priority Date: 2020-10-14
- Substituted dihydropyranopyrimidine compounds as kras inhibitorsPublication Number: WO-2022081655-A1Priority Date: 2020-10-14
- Oxygen-containing heterocyclic compound, preparation method and application thereofPublication Number: WO-2021109737-A1Priority Date: 2019-12-02
- Oxygen-containing heterocyclic compound, preparation method and application thereofPublication Number: EP-4015520-A1Priority Date: 2019-12-02



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
///////////talorasib, antineoplastic, 727W6T7DPK
It's only fair to share...