














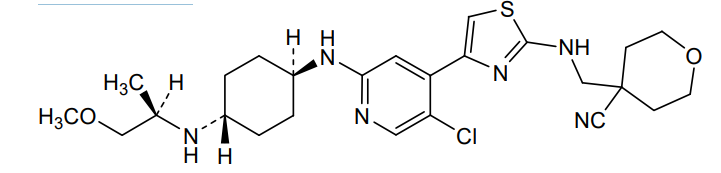
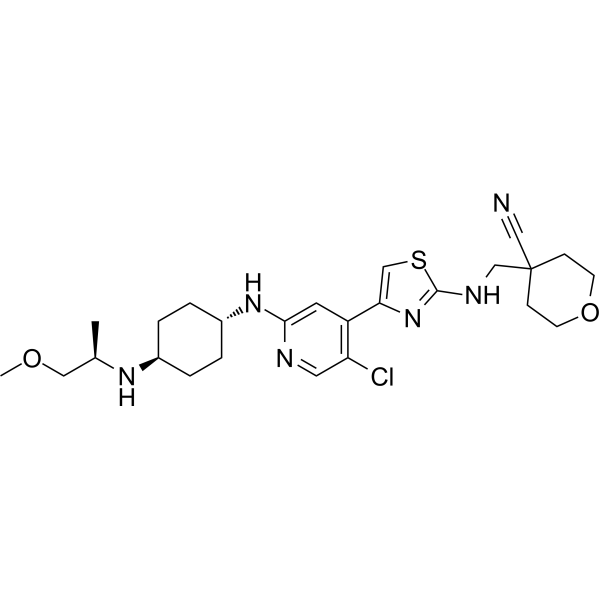
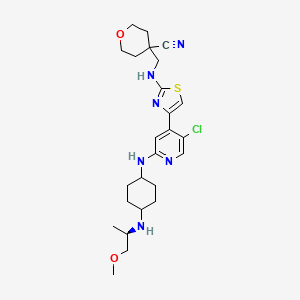
Tambiciclib
CAS 2247481-08-7
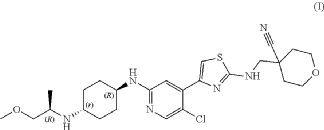
MF C25H35ClN6O2S, 519.10
4-[[[4-[5-chloro-2-[[4-[[(2R)-1-methoxypropan-2-yl]amino]cyclohexyl]amino]-4-pyridinyl]-1,3-thiazol-2-yl]amino]methyl]oxane-4-carbonitrile
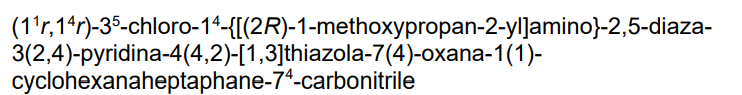
cyclin-dependent kinase inhibitor, antineoplastic, GFH 009, JSH 009, XDZ7VK8CXC, Orphan Drug , Acute myeloid leukaemia, Peripheral T-cell lymphoma
Tambiciclib (GFH009, JSH-009) is an orally active, highly potent and selective CDK9 inhibitor (IC50 = 1 nM), demonstrating >200-fold selectivity over other CDKs, >100-fold selectivity over DYRK1A/B, and excellent selectivity over 468 kinases/mutants. Tambiciclib demonstrates potent in vitro and in vivo antileukemic efficacy in acute myeloid leukemia (AML) mouse models by inhibiting RNA Pol II phosphorylation, downregulating MCL1 and MYC, and inducing apoptosis. Tambiciclib can be used for AML research.
Tambiciclib is a selective inhibitor of the serine/threonine cyclin-dependent kinase 9 (CDK9), the catalytic subunit of the RNA polymerase II (RNA Pol II) elongation factor positive transcription elongation factor b (PTEF-b; PTEFb), with potential antineoplastic activity. Upon administration, tambiciclib targets, binds to and blocks the phosphorylation and kinase activity of CDK9, thereby preventing PTEFb-mediated activation of RNA Pol II, leading to the inhibition of gene transcription of various anti-apoptotic proteins. This induces cell cycle arrest and apoptosis and prevents tumor cell proliferation. CDK9 regulates elongation of transcription through phosphorylation of RNA Pol II at serine 2 (p-Ser2-RNAPII). It is upregulated in various tumor cell types and plays a key role in the regulation of Pol II-mediated transcription of anti-apoptotic proteins. Tumor cells are dependent on anti-apoptotic proteins for their survival.
- OriginatorGenFleet Therapeutics
- DeveloperGenFleet Therapeutics; Sellas Life Sciences Group
- ClassAntineoplastics; Small molecules
- Mechanism of ActionCyclin-dependent kinase 9 inhibitors
- Orphan Drug StatusYes – Acute myeloid leukaemia; Peripheral T-cell lymphoma
- Phase IIAcute myeloid leukaemia
- Phase I/IIDiffuse large B cell lymphoma; Haematological malignancies; Peripheral T-cell lymphoma
- Phase ISolid tumours
- PreclinicalColorectal cancer; T-cell prolymphocytic leukaemia
- 13 Oct 2025Preclinical trials in T-cell prolymphocytic leukaemia (Combination therapy) in USA (Parenteral)
- 13 Oct 2025Preclinical trials in T-cell prolymphocytic leukaemia (Monotherapy) in USA (Parenteral)
- 13 Oct 2025Pharmacodynamics data from preclinical studies in T-cell prolymphocytic leukaemia released by SELLAS Life Sciences
CLINICAL
- A Study of GFH009 in Combination With Zanubrutinib in Subjects With Relapsed or Refractory DLBCLCTID: NCT06375733Phase: Phase 1/Phase 2Status: RecruitingDate: 2025-08-12
- A Study of GFH009 Monotherapy in Patients with Relapsed or Refractory Peripheral T-cell Lymphoma (PTCL)CTID: NCT05934513Phase: Phase 1/Phase 2Status: RecruitingDate: 2024-12-13
Publication Name: European Journal of Medicinal Chemistry
Publication Date: 2018-10-05
PMID: 30253346
DOI: 10.1016/j.ejmech.2018.09.025
SYN
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018192273&_cid=P12-MJ18VV-17351-1
Example 1: Synthesis of 4-(((4-(5-chloro-2-(((1R,4r)-4-(((R)-1-methoxypropyl-2-yl)amino) cyclohexyl)amino)pyridin-4-yl)thiazolyl)amino)methyl)tetrahydro-2H-pyran-4- carboxynitrile
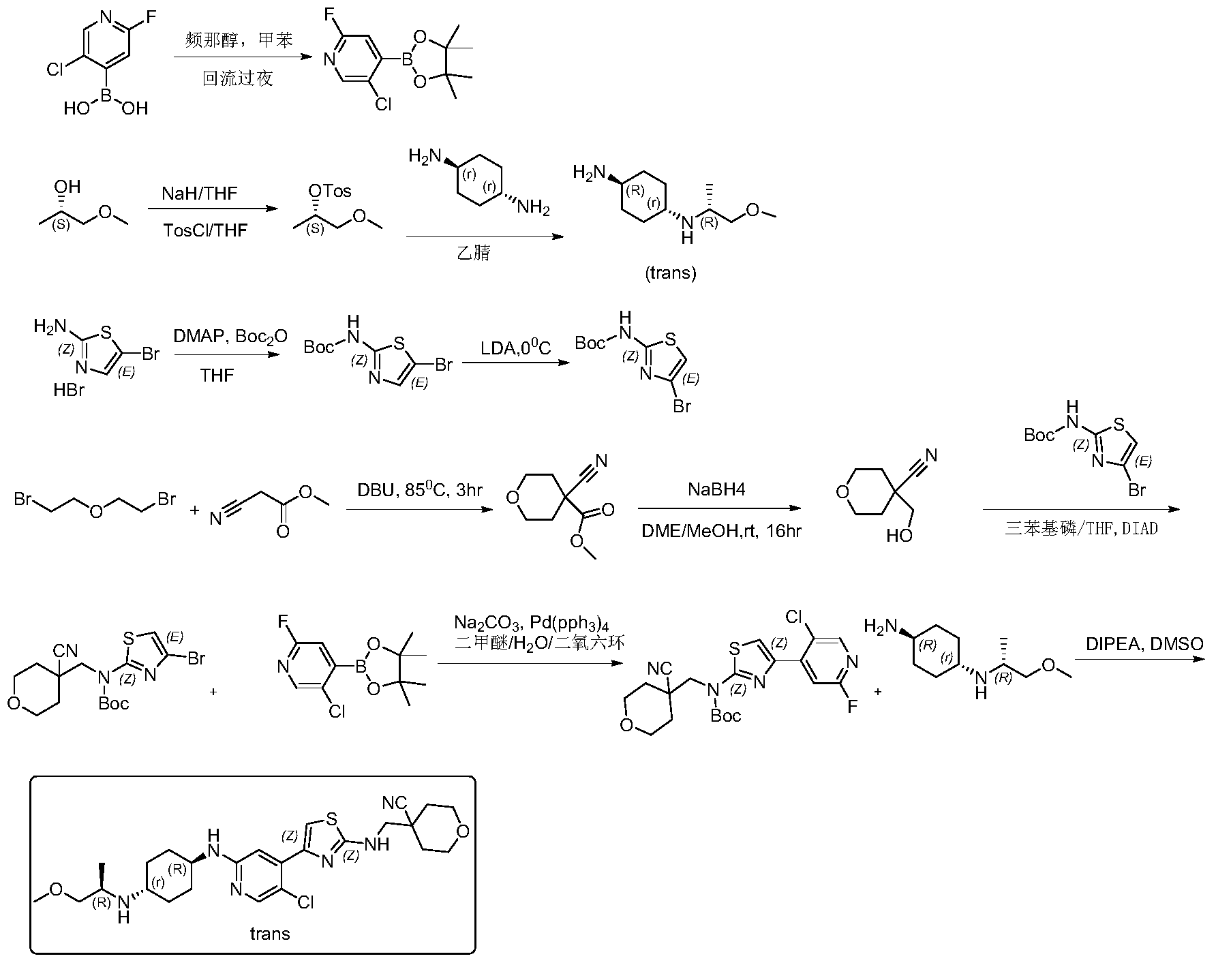
Step 1: Synthesis of 5-chloro-2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborhecyclopentan-2-yl)pyridine
[0102]5-Chloro-2-fluoropyridine-4-boronic acid (0.7 g, 4.46 mmol) and pinacol (0.63 g, 5.35 mmol) were added to 50 mL of toluene, and the mixture was refluxed at 120 °C overnight. TLC showed a small amount of starting material remaining. The reaction mixture was cooled to room temperature and concentrated, then dried by an oil pump to give 0.92 g of a white solid compound, 5-chloro-2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborhexacyclopentan-2-yl)pyridine, yield 80%, MS (ESI): m/z 258.1 (M+H) + .
[0103]Step 2: Synthesis of (S)-1-methoxypropyl-2-yl-4-toluenesulfonyl ester
[0104]60% sodium hydride (NaH) (6.52 g, 283 mmol) was added to anhydrous tetrahydrofuran (THF) (200 mL). The mixture was cooled to 0 °C in an ice bath under nitrogen protection, and (S)-(+)-1-methoxy-2-propanol (21 g, 233 mmol) was added dropwise. After the addition was complete, the mixture was brought to room temperature and stirred for 1.5 hours. The reaction mixture was then cooled back to 0 °C, and a tetrahydrofuran (THF) solution of p-toluenesulfonyl chloride (45.3 g, 283 mmol) (200 mL) was added dropwise. After the addition was complete, the mixture was stirred overnight at room temperature. TLC showed that the starting material had reacted completely. The reaction mixture was diluted with ethyl acetate (500 mL), and the reaction was quenched by adding water (500 mL) dropwise while cooling in an ice bath. The mixture was separated, and the aqueous phase was extracted once more with ethyl acetate (200 mL). The combined organic phases were washed with water (200 mL) and then with saturated brine (200 mL). The crude product was dried with anhydrous sodium sulfate, filtered, and concentrated to obtain 43 g of a pale yellow oily substance. Column separation (petroleum ether/ethyl acetate = 5/1) yielded 37 g of (S)-1-methoxypropyl-2-yl-4-toluenesulfonyl ester, a pale yellow oily substance, with a yield of 65.1%. MS (ESI): m/z 245.1 (M+H) + .
[0105]Step 3: Synthesis of (1r,4R)-N
1 -((R)-1-methoxypropyl-2-yl)cyclohexane-1,4-diamine
[0106](S)-1-methoxypropyl-2-yl 4-toluenesulfonyl ester (5 g, 20.5 mmol) and trans-1,4-cyclohexanediamine (5.84 g, 51.2 mmol) were added to 50 mL of acetonitrile and heated to 90 °C overnight. The reaction was monitored by TLC until complete. After cooling, the reaction solution was filtered, the filtrate was concentrated, and the residue was dissolved in dichloromethane and separated by silica gel stirring column (dichloromethane/methanol = 10/1) to give 2.5 g of the pale yellow liquid compound (1r,4R)-N
1 -((R)-1-methoxypropyl-2-yl)cyclohexane-1,4-diamine, yield 65%, MS (ESI): m/z 187.3 (M+H) + .
[0107]Step 4: Synthesis of tert-butyl 5-bromothiazol-2-ylcarbamate
[0108]105 g (403 mmol) of 5-bromothiazol-2-amine hydrobromide was suspended in 500 mL of tetrahydrofuran. Dimethylaminopyridine (2.41 g, 20 mmol) was added, resulting in a white turbidity. A tetrahydrofuran solution of di-tert-butyl dicarbonate (105.6 g, 484.6 mmol) was slowly added dropwise, and the reaction was allowed to proceed at room temperature for two days. The reaction solution was concentrated and dissolved in 300 mL of dichloromethane. The solution was mixed with silica gel and separated by column chromatography (petroleum ether/ethyl acetate = 10/1-6/1 gradient elution) to give 45 g of off-white solid, yield 40%. MS (ESI): m/z 278.98 (M+H) + .
[0109]Step 5: Synthesis of tert-butyl 4-bromothiazol-2-ylcarbamate
[0110]A 200 mL solution of diisopropylamine (64 mL, 446 mmol) in tetrahydrofuran was added to a dry three-necked flask. Under nitrogen protection, the mixture was cooled to 0 °C, and n-butyllithium (2.5 M, 173 mL, 431.7 mmol) was added dropwise. The reaction was allowed to proceed for 1 hour after the addition was complete. Then, a 400 mL solution of 5-bromothiazol-2-ylcarbamate in tetrahydrofuran was added dropwise at 0 °C. The reaction was allowed to proceed for 2 hours after the addition was complete. TLC showed that the reaction was complete. At 0℃, ice water (5 mL) was slowly added dropwise to quench the reaction. After stirring for 30 minutes, saturated ammonium chloride (500 mL) aqueous solution was added. The mixture was separated, and the aqueous layer was extracted with dichloromethane (2 × 300 mL). The organic layers were combined, washed with saturated brine, dried with anhydrous sodium sulfate, filtered, concentrated, and recrystallized from petroleum ether:ethyl acetate = 30:1. 31 g of tert-butyl 4-bromothiazol-2-ylcarbamate was obtained as a white solid, yield 77.5%. MS (ESI): m/z 278.98 (M+H) + .
[0111]Step Six: Synthesis of Methyl 4-cyano-tetrahydro-2H-pyran-4-carbonate
[0112]Methyl cyanoacetate (39.1 g, 395.3 mmol) and 2,2-dibromoethyl ether (100 g, 434.8 mmol) were added to 600 mL of dimethylformamide, followed by DBU (90 g, 593 mmol). The mixture was heated to 85 °C and reacted for 3 hours. TLC showed that the starting material reacted completely. The solid was filtered off, washed with ethyl acetate (2 × 300 mL), and the mother liquor was concentrated to obtain a brown oily substance. The oil was distilled under reduced pressure at an internal temperature of 65-70 °C, and the fraction collected was a colorless liquid. Crystallization was observed to give 42 g of a white solid, 4-cyano-tetrahydro-2H-pyran-4-carbonate. Yield: 62.8%, MS (ESI): m/z 178.2 (M+H) + .
[0113]Step 7: Synthesis of 4-(hydroxymethyl)-tetrahydro-2H-pyran-4-carboxynitrile
[0114]4-Cyano-tetrahydro-2H-pyran-4-carbonate methyl ester (42 g, 248.4 mmol) was dissolved in 400 mL of ethylene glycol dimethyl ether and 40 mL of methanol. The mixture was cooled to 0 °C in an ice bath, and sodium borohydride (11.1 g, 149 mmol) was added in portions. After the addition was complete, the mixture was allowed to rise to room temperature and stirred for 16 hours. The reaction was completed by TLC. The reaction solution was concentrated, and methanol was added to quench excess sodium borohydride. The solution was then concentrated again. Column chromatography (petroleum ether/ethyl acetate = 5/1) yielded 28 g of 4-(hydroxymethyl)-tetrahydro-2H-pyran-4-carboxynitrile, a pale yellow oil, yield: 79.5%, MS (ESI): m/z 142.1 (M+H) + .
[0115]Step 8: Synthesis of tert-butyl (4-bromothiazolyl)((4-cyanotetrahydro-2H-pyran-4-yl)methyl)carbamate
[0116]4-(hydroxymethyl)-tetrahydro-2H-pyran-4-carboxynitrile, 4-bromothiazol-2-ylcarbamate tert-butyl ester, and triphenylphosphine were added to anhydrous tetrahydrofuran (THF) and cooled to 0°C. Diisopropyl azodicarbonate (DIAD) was added dropwise. The mixture was stirred at room temperature for 10 minutes, then heated to 40°C overnight. The reaction solution was concentrated, and the residue was dissolved in dichloromethane. The solution was mixed with silica gel and separated by column chromatography (petroleum ether/ethyl acetate = 50/1, 30/1, 20/1) to obtain (4-bromothiazol-2-yl)((4-cyanotetrahydro-2H-pyran-4-yl)methyl)carbamate tert-butyl ester, a white solid of 365 mg, yield 50%. MS (ESI): m/z 402.1 (M+H) + .
[0117]Step Nine: Synthesis of tert-butyl (4-(5-chloro-2-fluoropyridin-4-yl)thiazolyl)((4-cyano-tetrahydro-2H-pyran-4-yl)methyl)carbamate
[0118]5-Chloro-2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborhexacyclopentan-2-yl)pyridine and sodium carbonate were added to a mixture of dimethyl ether/H₂O
/ dioxane. The system was purged with nitrogen twice. Then, tert-butyl (4-bromothiazolyl)((4-cyanotetrahydro-2H-pyran-4-yl)methyl)carbamate and tetraphenylphosphine palladium Pd(pph 3 )
4 were added . The system was purged with nitrogen three times. The temperature was then raised to 70°C and the reaction was carried out for 6 hours. TLC showed that only half of the starting material remained. Heating was then stopped and the reaction was terminated. The reaction solution was cooled to room temperature, ethyl acetate and methanol were added, and the mixture was filtered. The filter cake was washed with ethyl acetate, the filtrate was concentrated, and the residue was dissolved in dichloromethane. The residue was washed with saturated brine, separated, and the organic phase was dried over anhydrous sodium sulfate. The mixture was filtered, and silica gel was added for mixing. The sample was separated by column chromatography (petroleum ether/ethyl acetate = 30/1) to give 3.2 g of (4-(5-chloro-2-fluoropyridin-4-yl)thiazolyl)((4-cyano-tetrahydro-2H-pyran-4-yl)methyl)carbamate, a white foamy solid, with a yield of 55%. MS (ESI): m/z 453.1 (M+H) + .
[0119]Step 10: Synthesis of 4-(((4-(5-chloro-2-(((1R,4r)-4-(((R)-1-methoxypropyl-2-yl)amino)cyclohexyl)amino)pyridin-4-yl)thiazolyl)amino)methyl)tetrahydro-2H-pyran-4-carboxynitrile
[0120]The tert-butyl carbamate (4-(5-chloro-2-fluoropyridin-4-yl)thiazolyl)((4-cyano-tetrahydro-2H-pyran-4-yl)methyl)carbamate (3.2 g, 7.1 mmol) and (1r,4R)-N
1 -((R)-1-methoxypropyl-2-yl)cyclohexane-1,4-diamine (3.9 g, 21.2 mmol) and diisopropylethylamine (DIPEA) were added to 30 mL of dimethyl sulfoxide. Under nitrogen protection, the mixture was heated to 100-110 °C and reacted for two days. The reaction was monitored by TLC and LCMS. The starting material (4-(5-chloro-2-fluoropyridin-4-yl)thiazolyl)((4-cyano-tetrahydro-2H-pyran-4-yl)methyl)carbamate tert-butyl ester had completely disappeared, with some BOC-free intermediate remaining. The reaction was stopped, and the reaction solution was cooled and diluted with ethyl acetate (60 mL). Water (150 mL) was added under ice bath. The mixture was separated, and the aqueous layer was extracted again with ethyl acetate (2 × 50 mL). The organic layers were combined, washed with saturated brine (100 mL), dried with anhydrous sodium sulfate, filtered, and concentrated to obtain a crude product of yellowish-brown oil. Column separation (acetonitrile/water/trifluoroacetic acid = 80/20/0.001) yielded 700 mg of 4-(((4-(5-chloro-2-(((1R,4r)-4-(((R)-1-methoxypropyl-2-yl)amino)cyclohexyl)amino)pyridin-4-yl)thiazolyl)amino)methyl)tetrahydro-2H-pyran-4-carboxynitrile, a pale yellow solid. Yield: 19.1%. ¹H NMR (400 MHz, CDCl₃
) )δ8.06(s,1H),7.38(s,1H),6.97(s,1H),5.92(brs,1H),4.45(d,J=8.0Hz,1H),4.02(dd,J 1=2.8Hz, J2=12Hz,2H),3.71-3.74(m,4H),3.54-3.56(m,1H),3.35(s,3H),3.21-3.25(m,2 H),3.00-3.05(m,1H),2.50-2.60(m,1H),2.15(d,J=9.6Hz,2H),2.04-2.07(m,1H),1.95(d ,J=12.8Hz,3H),1.74-1.82(m,3H),1.10-1.30(m,4H),1.00(d,J=.4Hz,3H),MS(ESI):m/z 519.3(M+H) + .
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US376039987&_cid=P12-MJ18R0-12787-1
PAT
- A novel cyclin-dependent kinase CDK9 inhibitorPublication Number: CN-108727363-BPriority Date: 2017-04-19Grant Date: 2020-06-19
- Inhibitor of cyclin-dependent kinase CDK9Publication Number: US-10952999-B2Priority Date: 2017-04-19Grant Date: 2021-03-23
- Novel inhibitor of cyclin-dependent kinase cdk9Publication Number: EP-3613737-B1Priority Date: 2017-04-19Grant Date: 2021-12-29
- Pharmaceutical combination and use thereof in treatment of cancerPublication Number: WO-2024239512-A1Priority Date: 2023-05-22
- Polymorph of cdk9 inhibitor and preparation method for polymorph and use thereofPublication Number: WO-2020244612-A1Priority Date: 2019-06-06
- Polymorphic substance of CDK9 inhibitor and preparation method and application thereofPublication Number: CN-113966332-APriority Date: 2019-06-06
- Novel inhibitor of cyclin-dependent kinase cdk9Publication Number: EP-3613737-A1Priority Date: 2017-04-19
- Novel inhibitor of cyclin-dependent kinase cdk9Publication Number: US-2020078343-A1Priority Date: 2017-04-19



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
//Tambiciclib, cyclin-dependent kinase inhibitor, antineoplastic, GFH 009, JSH 009, XDZ7VK8CXC, Orphan Drug , Acute myeloid leukaemia, Peripheral T-cell lymphoma