














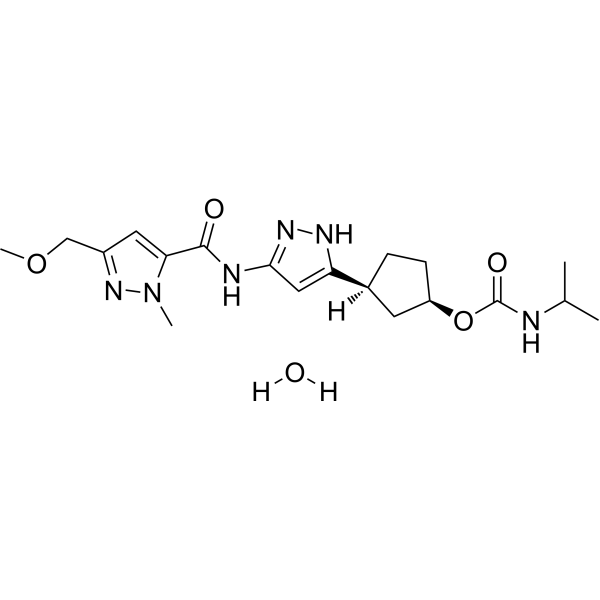
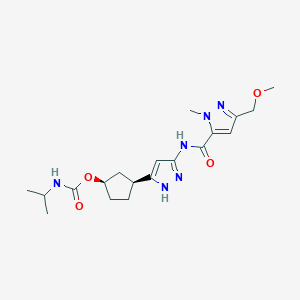
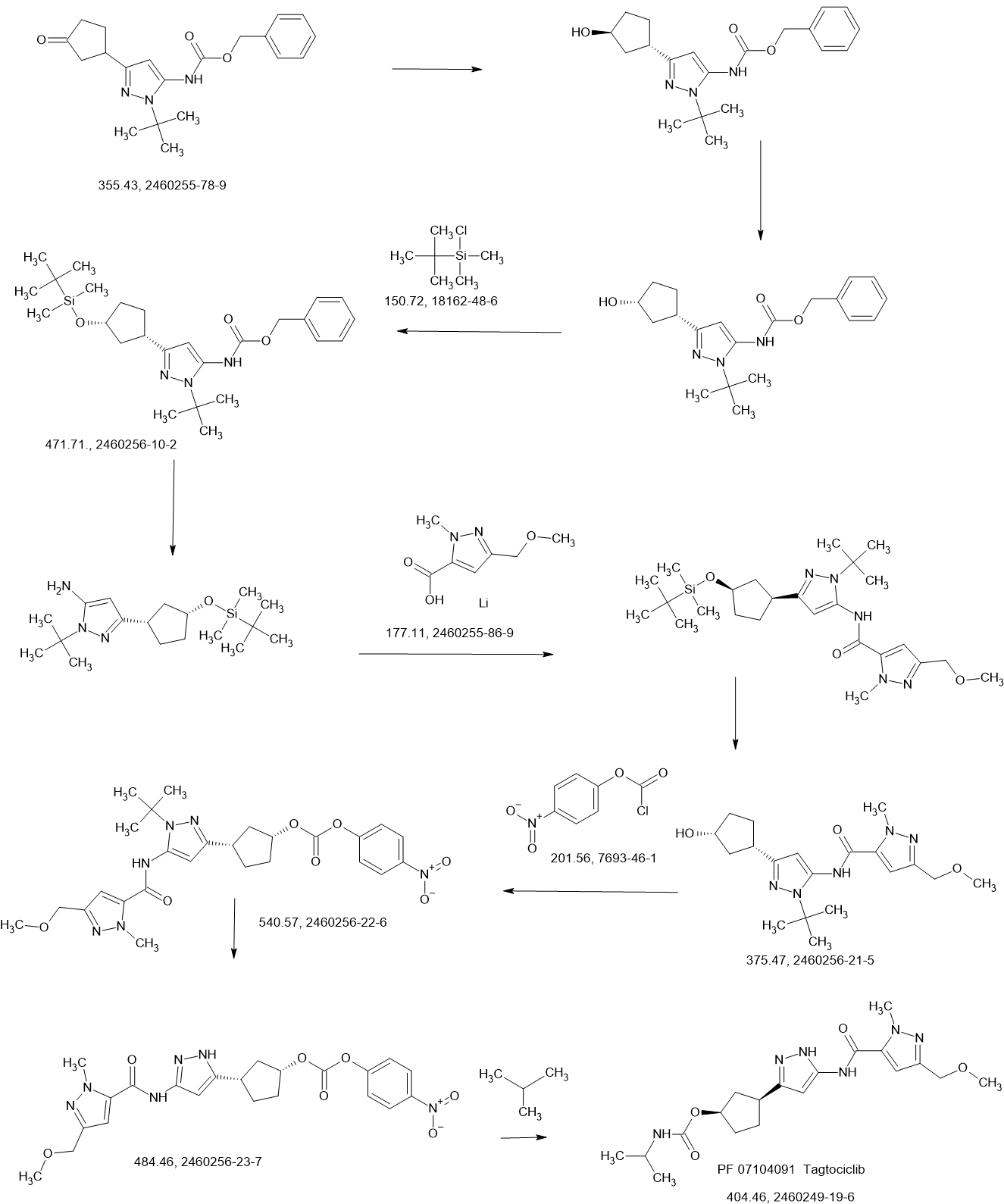
Tagtociclib (PF-07104091), 2460249-19-6, MW 404.5, C19H28N6O4
CAS 2733575-91-0 HYDRATE
| Molecular Weight HYDRATE | 422.48 |
|---|---|
| Formula | C19H30N6O5 |
[(1R,3S)-3-[3-[[5-(methoxymethyl)-2-methylpyrazole-3-carbonyl]amino]-1H-pyrazol-5-yl]cyclopentyl] N-propan-2-ylcarbamate
- (1R,3S)-3-[5-[[[3-(Methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl]amino]-1H-pyrazol-3-yl]cyclopentyl N-(1-methylethyl)carbamate
- (1R,3S)-3-{5-[3-(methoxymethyl)-1-methyl-1H-pyrazole-5carboxamido]-1H-pyrazol-3-yl}cyclopentyl (propan-2yl)carbamate
- (1R,3S)-3-(3-(3-(Methoxymethyl)-1-methyl-1H-pyrazole-5-carboxamido)-1H-pyrazol-5-yl)cyclopentylisopropylcarbamate
- Carbamic acid, N-(1-methylethyl)-, (1R,3S)-3-[5-[[[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl]amino]-1H-pyrazol-3-yl]cyclopentyl ester
PF-07104091 hydrate is a potent and selective CDK2/cyclin E1 and GSK3β inhibitor, with Kis of 1.16 and 537.81 nM, respectively. PF-07104091 hydrate has anti-tumor activity for cyclin E1-amplified cancers. (patent WO2020157652A2).
- OriginatorPfizer
- ClassAntineoplastics; Small molecules
- Mechanism of ActionCyclin-dependent kinase 2 inhibitors
Phase IIBreast cancer; Solid tumours
Phase I/IINon-small cell lung cancer; Ovarian cancer; Small cell lung cancer
13 Sep 2024Efficacy, adverse events, pkarmacokinetics and pharmacodynamics data from a phase I/II trial in Solid tumours presented at the 49th European Society for Medical Oncology Congress (ESMO-2024)
13 Sep 2024Pharmacodynamics data from a preclinical trial in Breast cancer presented at the 49th European Society for Medical Oncology Congress (ESMO-2024)
05 Apr 2024Pharmacodynamics data form preclinical trial in Breast cancer and Ovarian cancer presented at the 115th Annual Meeting of the American Association for Cancer Research (AACR-2024)
Tegtociclib is an orally bioavailable inhibitor of cyclin-dependent kinase 2 (CDK2), with potential antineoplastic activity. Upon administration, tegtociclib selectively targets, binds to and inhibits the activity of CDK2. This may lead to cell cycle arrest, the induction of apoptosis, and the inhibition of tumor cell proliferation. CDKs are serine/threonine kinases that are important regulators of cell cycle progression and cellular proliferation and are frequently overexpressed in tumor cells. CDK2/cyclin E complex plays an important role in retinoblastoma (Rb) protein phosphorylation and the G1-S phase cell cycle transition. CDK2/cyclin A complex plays an important role in DNA synthesis in S phase and the activation of CDK1/cyclin B for the G2-M phase cell cycle transition.
SCHEME
COUPLER
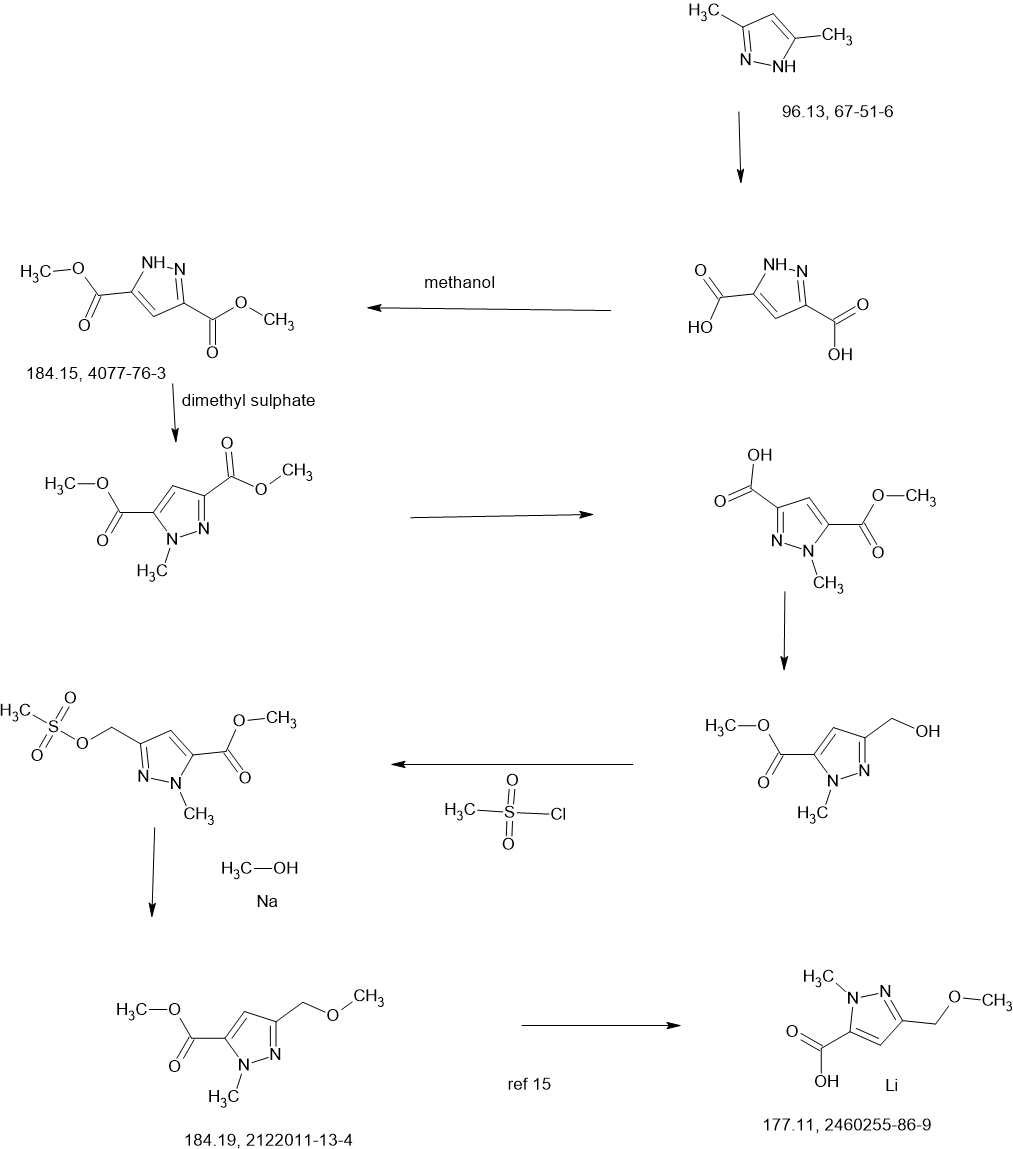
MAIN
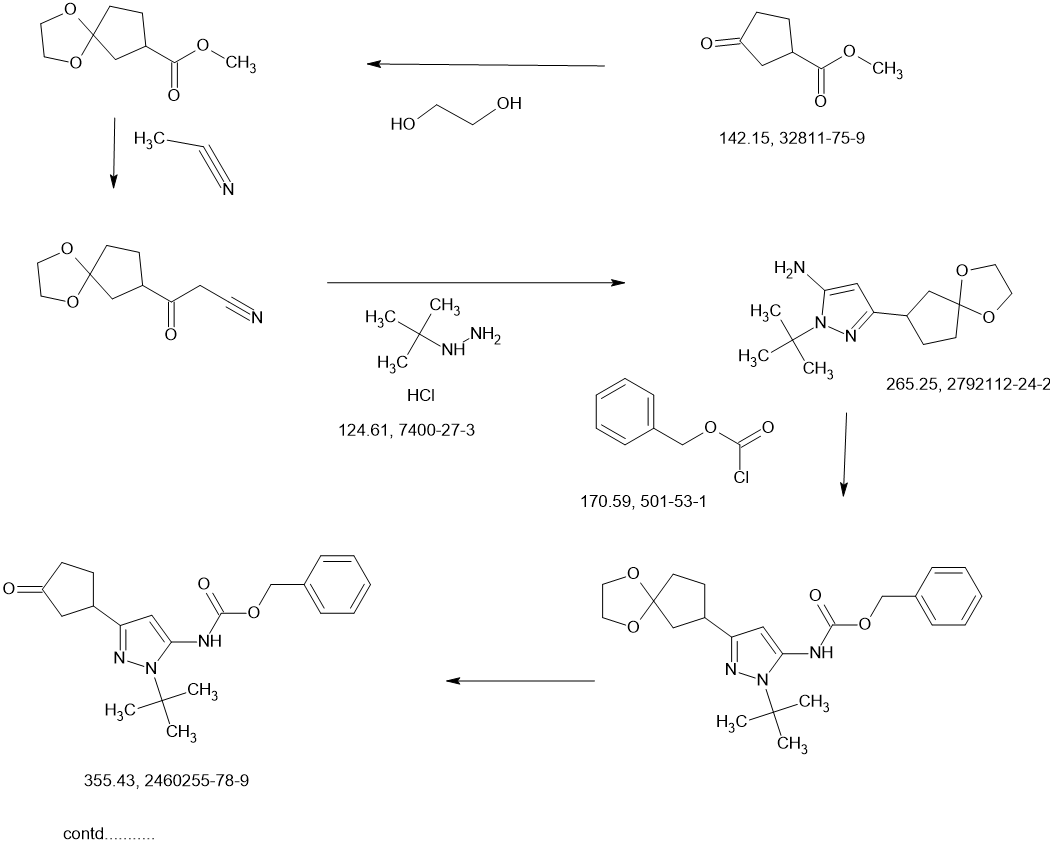
CONTD………….
PATENTS
WO2022018596 78%
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022018596&_cid=P22-MDFCVG-44044-1
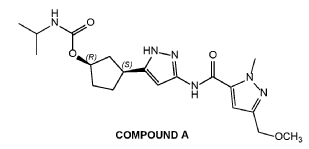
COMPOUND A was prepared as described in Example 13 of U.S. Patent No.
11,014,911.
Preparation of Intermediate 1: benzyl {1-tert-butyl-3-[(1S,3R)-3-hvdroxycvclopentyl]1H-pyrazol-5-yl)carbamate; and Intermediate 2: benzyl {1-tert-butyl-3-[(1R,3S)-3-hydroxycvclopentyl1-1H-pyrazol-5-yl)carbamate.
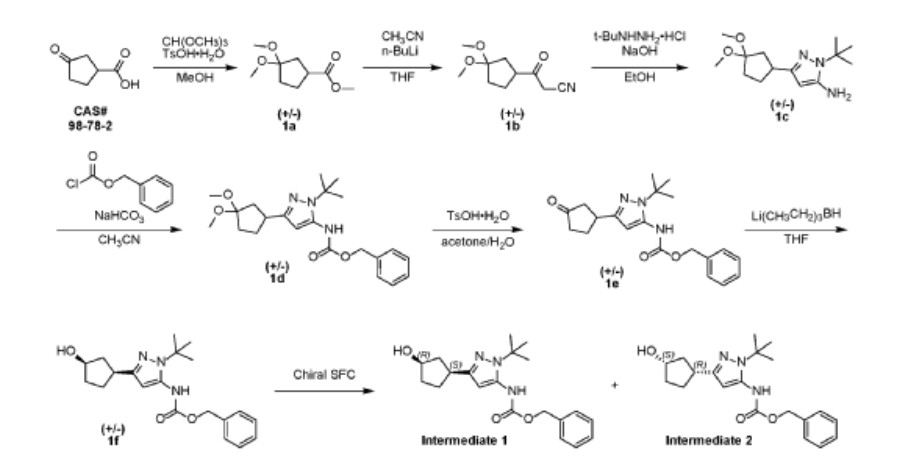
Two parallel reactions, each containing a solution of (±)-3- oxocyclopentanecarboxylic acid (CAS#98-78-2, 900 g, 7.02 mol) in methanol (5 L) at 13 °C were each treated with trimethyl orthoformate (4.47 kg, 42.15 mol, 4.62 L) and 4- toluenesulfonic acid monohydrate (26.72 g, 140.5 mmol). The mixtures were stirred at 13 °C for 25 hours. Each batch was quenched separately with sat. aq NaHCO3 (1 L), then the two batches were combined and concentrated under vacuum to remove most of the methanol. The residue was diluted with ethyl acetate (4 L), and the layers separated. The aqueous layer was further extracted with ethyl acetate (2 x 1 L). The combined organic layers were washed with sat. aq NaCI (3 x 1 L), dried over magnesium sulfate, filtered, and concentrated under vacuum to give (±)-methyl 3,3- dimethoxycyclopentanecarboxylate (1a, 2.5 kg, 13.28 mol, 94%) as a light yellow oil. 1H NMR (400MHz, CHLOROFORM -d) δ = 3.67 (s, 3H), 3.20 (s, 3H), 3.19 (s, 3H), 2.94- 2.82 (m, 1 H), 2.16-2.00 (m, 2H), 1.99-1.76 (m, 4H).
A solution of n-butyllithium (3.44 L of a 2.5 M solution in hexanes, 8.6 mol) was added to a reactor containing THF (3 L) at -65 °C. Anhydrous acetonitrile (453 mL, 353 g, 8.61 mol) was added dropwise, slowly enough to maintain the internal temperature below -55 °C. The mixture was stirred for an additional 1 hour at -65 °C. A solution of (±)-methyl 3,3-dimethoxycyclopentanecarboxylate (1a, 810 g, 4.30 mol) in THF (1 L) was then added dropwise, slowly enough to maintain the internal temperature below -50 °C. After stirring for an additional hour at -65 °C, the reaction was quenched with water (4 L), neutralized with aq HCI (1 M) to pH 7, and extracted with ethyl acetate (3 x 3L). The combined organic layers were washed with sat. aq NaCI (2 x 3L), dried over magnesium sulfate, filtered, and concentrated under vacuum to give crude (±)-3-(3,3-dimethoxycyclopentyl)-3-oxopropanenitrile (1b, 722 g, 3.66 mol, 85%) as a red oil, which was used without further purification.
Solid sodium hydroxide (131.4 g, 3.29 mol total) was added in portions to a suspension of tert-butylhydrazine hydrochloride (409.4 g, 3.29 mol) in ethanol (3 L) at 16-25 °C. Stirring was continued at 25 °C for 1 hour. A solution of crude (±)-3-(3,3-dimethoxycyclopentyl)-3-oxopropanenitrile (1b, 540 g, 2.74 mol) in ethanol was added at 25 °C, then the mixture was heated to 75 °C internal and stirred for 30 hours. The reaction was filtered, and the filtrate concentrated under vacuum to give crude product as a red oil. This product was combined with crude from three more identically-prepared batches (each starting with 540 g 1b; 2.16 kg, 10.96 mol total for the 4 batches), and purified by silica gel chromatography (eluting with 0-35% ethyl acetate in petroleum ether), affording (±)-1-tert-butyl-3-(3,3-dimethoxycyclopentyl)-1H-pyrazol-5-amine (1c, 1.60 kg, 5.98 mol, 54% yield) as a red oil. 1H NMR (CHLOROFORM -d) δ = 5.41 (s, 1 H), 3.50 (br. s., 2H), 3.22 (s, 3H), 3.20 (s, 3H), 3.13 (tt, J=7.9, 9.6 Hz, 1H), 2.25 (dd, J=8.0, 13.3 Hz, 1H), 2.09-2.00 (m, 1H), 1.99-1.91 (m, 1H), 1.83 (dd, J=10.8, 12.8 Hz, 2H), 1.78-1.68 (m, 1H), 1.60 (s, 9H).
Benzyl chloroformate (563.6 mL, 676.3 g, 3.96 mol) was added to a chilled (0-5 °C) solution of (±)-1-tert-butyl-3-(3,3-dimethoxycyclopentyl)-1H-pyrazol-5-amine (1c, 530 g, 1.98 mol) in acetonitrile (3.5 L). The mixture was stirred at 23 °C for 2 hours, and then solid sodium hydrogen carbonate (532.9 g, 6.34 mol) was added in portions. Stirring was continued at 23 °C for 26 hours. The resulting suspension was filtered and the filtrate concentrated under vacuum to give crude (±)-benzyl [1-tert-butyl-3-(3,3-dimethoxycyclopentyl)-1H-pyrazol-5-yl]carbamate (1 d, 980 g, 1.98 mol max) as a red oil, which was used in the next step without further purification.
A solution of the crude (±)-benzyl [1-tert-butyl-3-(3,3-dimethoxycyclopentyl)-1H-pyrazol-5-yl]carbamate (1 d, 980 g, 1.98 mol max) in acetone (2 L) and water (2 L) at 18 °C was treated with 4-toluenesulfonic acid monohydrate (48.75 g, 256.3 mmol). The mixture was heated to 60 °C internal for 20 hours. After concentration under vacuum to remove most of the acetone, the aqueous residue was extracted with dichloromethane (3 x 3 L). The combined organic extracts were dried over sodium sulfate, filtered, and concentrated under vacuum to a crude red oil. This crude product was combined with crude from two other identically-prepared batches (each derived from 1.98 mol 1c, 5.94 mol total for the 3 batches), and purified by silica gel chromatography (eluting with 0- 50% ethyl acetate in petroleum ether) to give (±)-benzyl [1-tert-butyl-3-(3-oxocyclopentyl)-1H-pyrazol-5-yl]carbamate (1 e, 1.6 kg) as a yellow solid. This solid was stirred in 10:1 petroleum ether/ethyl acetate (1.5 L) at 20 °C for 18 hours. The resulting suspension was filtered, the filter cake washed with petroleum ether ( 2 x 500 mL), and the solids dried under vacuum to give (±)-benzyl [1-tert-butyl-3-(3-oxocyclopentyl)-1H-pyrazol-5-yl]carbamate (1 e, 1.4 kg, 3.9 mol, 66% combined for the three batches). 1H NMR (DMSO–d6) δ = 9.12 (br. s., 1H), 7.56-7.13 (m, 5H), 6.03 (s, 1 H), 5.12 (s, 2H), 3.41-3.27 (m, 1H), 2.48-2.39 (m, 1H), 2.34-2.10 (m, 4H), 1.98-1.81 (m, 1 H), 1.48 (s, 9H).
A solution of (±)-benzyl [1-tert-butyl-3-(3-oxocyclopentyl)-1H-pyrazol-5-yl]carbamate (1 e, 320 g, 0.900 mol) in THF (1.5 L) was degassed under vacuum and purged with dry nitrogen (3 cycles), then cooled to -65 °C internal. A solution of lithium triethylborohydride (1.0 M in THF, 1.80 L, 1.80 mol) was added dropwise at a rate which maintained the internal temperature below -55 °C, then stirring was continued at -65 °C for 1.5 hours. The reaction mixture was quenched with sat. aq NaHCO3 (1.5 L) at -40 to -30 °C. Hydrogen peroxide (30% aqueous, 700 g) was added to the mixture dropwise, while the internal temperature was maintained at -10 to 0 °C. The mixture was stirred at 10 °C for 1 hour, then extracted with ethyl acetate (3 x 2 L). The combined organic layers were washed with sat. aq Na2SO3 (2 x 1 L) and sat. aq NaCI (2 x 1 L). The organics were dried over magnesium sulfate, filtered, and concentrated under vacuum to a crude yellow oil. The crude product from this batch was combined with crude from three other, identically-prepared batches (each starting from 0.900 mol 1 e, for a total of 3.60 mol) for purification. Before chromatography, the combined mixture showed ~3.3:1 cis/trans ratio by NMR. The combined crude product was purified twice by silica gel chromatography, eluting with 0-50% ethyl acetate in dichloromethane), affording (±)-trans-benzyl [1-tert-butyl-3-(3-hydroxycyclopentyl)-1H-pyrazol-5-yl]carbamate (1 f, 960 g) as a light yellow solid, which was further purified by trituration, as described below.
A previous batch of 1f had been obtained from smaller-scale reactions, starting from a total of 120 g 1e (0.34 mol). The columned product from this batch was combined with the columned product from the batch above (which had been derived from 3.60 mol 1 e, for a total of 3.94 mol 1e used for all the combined batches), suspended in 10:1 dichloromethane/methanol (1.5 L), and stirred at 20 °C for 16 hours. The suspension was filtered, and the filter cake washed with petroleum ether (2 x 500 mL). The solids were dried under vacuum to give clean (±)-trans-benzyl [1-tert-butyl-3-(3-hydroxycyclopentyl)-1H-pyrazol-5-yl]carbamate (1 f, 840 g, 2.35 mol, 60% total yield for all the combined batches) as a white solid. 1H NMR (400MHz, DMSO-d6) δ = 9.07 (br. s., 1 H), 7.45-7.27 (m, 5H), 5.92 (s, 1 H), 5.11 (s, 2H), 4.57 (d, J=4.5 Hz, 1 H), 4.21-4.07 (m, 1 H), 2.88 (quin, J=8.6 Hz, 1 H), 2.24-2.13 (m, 1 H), 1.92-1.78 (m, 1 H), 1.78-1.62 (m, 2H), 1.61-1.53 (m, 1 H), 1.47 (s, 9H), 1.52-1.43 (m, 1 H). MS: 358 [M+H]+.
The enantiomers of (±)-trans-benzyl [1-tert-butyl-3-(3-hydroxycyclopentyl)-1H-pyrazol-5-yl]carbamate (1 f, 700 g, 1.96 mol) were separated by chiral SFC.
The product from the first-eluting enantiomer peak (310 g solid) was suspended in methanol/petroleum ether (1 :10, 1 L) and stirred at 25 °C for 1 hour. The suspension was filtered, the filter pad washed with petroleum ether (2 x 500 mL), and the solids dried under vacuum to give benzyl {1-tert-butyl-3-[(1S,3R)-3-hydroxycyclopentyl]-1H-pyrazol-5-yl}carbamate (Intermediate 1 , 255 g, 713 mmol, 36%, >99% ee) as a white solid. 1H NMR (400MHz, DMSO -d6) δ = 9.08 (br. s., 1 H), 7.58-7.20 (m, 5H), 5.92 (s, 1 H), 5.11 (s, 2H), 4.57 (d, J=4.4 Hz, 1 H), 4.19-4.09 (m, 1 H), 2.88 (quin, J=8.6 Hz, 1 H), 2.24-2.13 (m, 1 H), 1.91-1.79 (m, 1 H), 1.79-1.61 (m, 2H), 1.61-1.53 (m, 1 H), 1.47 (s, 9H), 1.52-1.44 (m, 1 H). MS: 358 [M+H]+. Optical rotation [α]D +3.76 (c 1.0, MeOH). Chiral purity: >99% ee, retention time 3.371 min. Chiral SFC analysis was performed on a ChiralPak AD-3 150 x 4.6 mm ID, 3 pm column heated to 40 °C, eluted with a mobile phase of CO2 and a gradient of 0-40% methanol+0.05%DEA over 5.5 min, then held at 40% for 3 min; flowing at 2.5 mL/min.
The product from the second-eluting enantiomer peak (300 g solid) was suspended in methanol/petroleum ether (1 :10, 1 L) and stirred at 25 °C for 1 hour. The suspension was filtered, the filter pad washed with petroleum ether (2 x 500 mL), and the solids dried under vacuum to give benzyl {1-tert-butyl-3-[(1R,3S)-3-hydroxycyclopentyl]-1H-pyrazol-5-yl}carbamate (Intermediate 2, 255 g, 713 mmol, 36%, 94% ee) as a white solid. 1H NMR (400MHz, DMSO-d6) δ = 9.08 (br. s., 1 H), 7.55-7.19 (m, 5H), 5.92 (s, 1 H), 5.11 (s, 2H), 4.57 (d, J=4.4 Hz, 1 H), 4.23-4.07 (m, 1 H), 2.88 (quin, J=8.7 Hz, 1 H), 2.23-2.14 (m, 1 H), 1.90-1.79 (m, 1 H), 1.77-1.61 (m, 2H), 1.61-1.53 (m, 1 H), 1 .47 (s, 9H), 1.52-1 .44 (m, 1 H). MS: 358 [M+H]+. Optical rotation [α]D -2.43 (c 1 .0, MeOH). Chiral purity: 94% ee, retention time 3.608 min. Chiral SFC analysis was performed on a ChiralPak AD-3 150 x 4.6 mm ID, 3 pm column heated to 40 °C, eluted with a mobile phase of CO2 and a gradient of 0-40% methanol+0.05%DEA over 5.5 min, then held at 40% for 3 min; flowing at 2.5 mL/min.
A sample of the second-eluting enantiomer from a previous batch with [α]D -3.1 (c 1.1, MeOH) and 96% ee was crystalized from dichloroethane/pentane. A crystal structure was obtained by small-molecule X-ray crystallography, which showed (1R,3S) geometry. The absolute stereochemistry of Intermediate 2 was thus assigned (1R,3S) based on its comparable optical rotation and order of elution in the analytical method. Intermediate 1, the enantiomer of Intermediate 2, was thus assigned (1S,3R) stereochemistry.
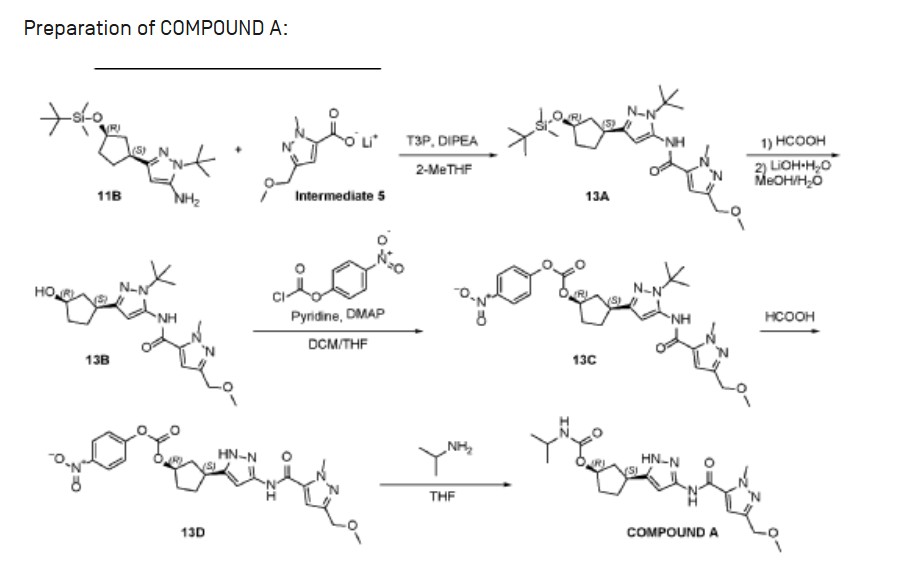
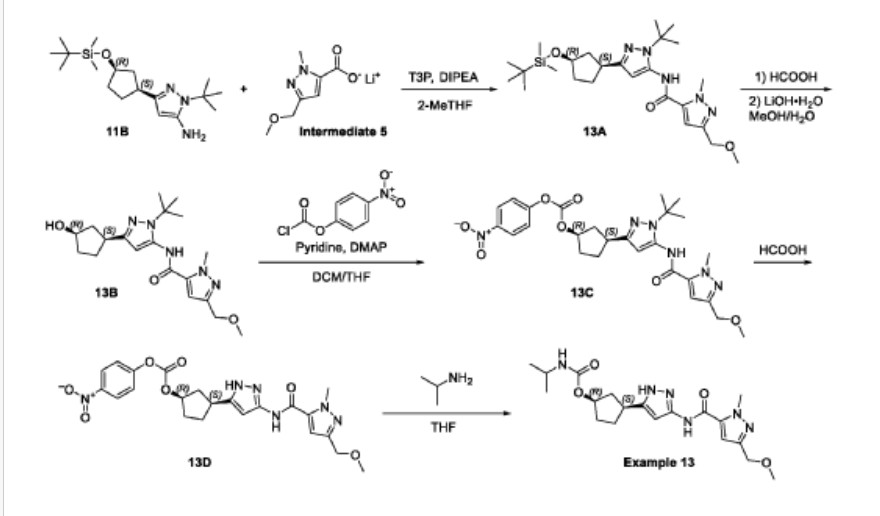
Propylphosphonic anhydride (T3P®, 50 wt% solution in EtOAc, 50.3 g, 79.1 mmol) was added to a room temperature (26 °C) solution of 1-tert-butyl-3-[(1S,3R)-3-{[tert-butyl(dimethyl)silyl]oxy}cyclopentyl]-1H-pyrazol-5-amine (11 B, 8.90g, 26.4 mmol), lithium 3-(methoxymethyl)-1-methyl-1H-pyrazole-5-carboxylate (Intermediate 5, 5.83 g,
34.3 mmol), and diisopropylethyl amine (10.2 g, 79.1 mmol) in 2-methyltetrahydrofuran (100.0 mL). The resulting mixture was stirred at this temperature for 18 hours. After concentrating the mixture to dryness, the residue was dissolved in dichloromethane (150 mL), and the solution washed sequentially with water (2 x 30 mL), sat. aq NaHCO3 (2 x 30 mL) and sat. aq NaCI (30 mL). The organic layer was dried over sodium sulfate, filtered, and concentrated to give crude N-{1-tert-butyl-3-[(1S,3R)-3-{[tert- butyl(dimethyl)silyl]oxy}cyclopentyl]-1H-pyrazol-5-yl}-3-(methoxymethyl)-1-methyl-1H- pyrazole-5-carboxamide (13A, 12.9 g, 100%) as an oil. MS: 490 [M+H]+.
The crude N-{1-tert-butyl-3-[(1S,3R)-3-{[tert-butyl(dimethyl)silyl]oxy}cyclopentyl]- 1H-pyrazol-5-yl}-3-(methoxymethyl)-1-methyl-1H-pyrazole-5-carboxamide (13A, 12.9 g,
26.3 mmol) was dissolved in formic acid (80 mL) and stirred at room temperature (27 °C) for 30 minutes. The mixture was concentrated to dryness, and the residue
dissolved in methanol (80 mL). A solution of lithium hydroxide monohydrate (3.43 g, 81.8 mmol) in water (15 mL) was added, and the mixture stirred at room temperature (27 °C) for 1 hour. The mixture was concentrated to dryness, and the residue was purified by silica gel chromatography (eluting with 0-80% ethyl acetate in petroleum ether) to give N-{1-tert-butyl-3-[(1S,3R)-3-hydroxycyclopentyl]-1H-pyrazol-5-yl}-3-(methoxymethyl)-1-methyl-1H-pyrazole-5-carboxamide (13B, 8.0 g, 78%) as a yellow gum. MS: 376 [M+H]+.
A solution of N-{1-tert-butyl-3-[(1S,3R)-3-hydroxycyclopentyl]-1H-pyrazol-5-yl}-3-(methoxymethyl)-1-methyl-1H-pyrazole-5-carboxamide (13B, 8.0 g, 21 mmol) in dichloromethane (80 mL) and THF (80 mL) was treated with DMAP (260 mg, 2.13 mmol), pyridine (5.06 g, 63.9 mmol), and 4-nitrophenyl chloroformate (8.59 g, 42.6 mmol). The resulting yellow suspension was stirred at room temperature for 18 hours. The reaction mixture was concentrated to dryness and purified by silica gel chromatography (eluting with 0-45% ethyl acetate in petroleum ether) to give (1R,3S)-3-[1-tert-butyl-5-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-3-yl]cyclopentyl 4-nitrophenyl carbonate (13C, 10.6 g, 92%) as a light brown gum. MS: 541 [M+H]+.
A solution of (1R,3S)-3-[1-tert-butyl-5-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-3-yl]cyclopentyl 4-nitrophenyl carbonate (13C, 10.6 g, 19.6 mmol) in formic acid (80 mL) was stirred at 70 °C for 18 hours. The solution was concentrated to dryness. The residue was dissolved in dichloromethane (150 mL) and the solution neutralized with sat. aq NaHCO3. The organic layer was washed with water (30 mL) and sat. aq NaCI (30 mL), dried over sodium carbonate, filtered, and concentrated to give crude (1R,3S)-3-[3-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-5-yl]cyclopentyl 4-nitrophenyl carbonate (13D, 8.5 g, 90%, 86% pure by LCMS) as a light yellow glass. MS: 485 [M+H]+.
A room temperature (27 °C) solution of crude (1R,3S)-3-[3-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-5-yl]cyclopentyl 4-nitrophenyl carbonate (13D, 1.7 g, 3.5 mmol) and 2-propylamine (1.04 g, 17.5 mmol) in THF (30 mL) was stirred for 6 hours. The solution was concentrated to dryness, and the residue was combined with the residue from a second batch which had been derived from 1.7 g, 3.5 mmol 13D (total 6.27 mmol 13D consumed for the combined two batches) to give 3.2 g crude product. This product was purified by preparative HPLC on a Phenomenex Gemini C18 250*50mm*10 pm column, eluting with 15-45% water (0.05% ammonium
hydroxide v/v) in acetonitrile. After lyophilization, (1R,3S)-3-[3-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-5-yl]cyclopentyl propan-2 -ylcarbamate (COMPOUND A, 2.06 g, 78%) was obtained as a white crystalline solid monohydrate. MS: 405 [M+H]+. 1H NMR (400MHz, DMSO-d6) d = 12.23 (br s, 1H), 10.73 (br s, 1H), 7.11 (s, 1H), 6.96 (br d, J=7.0 Hz, 1H), 6.41 (br s, 1H), 5.00 (br s, 1H), 4.33 (s, 2H), 4.04 (s, 3H), 3.57 (qd, J=6.6, 13.4 Hz, 1H), 3.26 (s, 3H), 3.17-2.96 (m, 1H), 2.48-2.39 (m, 1H), 2.03 (br d, J=6.8 Hz, 1H), 1.95-1.83 (m, 1H), 1.73 (br d, J=8.5 Hz, 2H), 1.61 (br s, 1 H), 1.03 (br d, J=6.3 Hz, 6H). Optical rotation [α]D +4.8 (c 1.0, MeOH). Chiral purity: >99% ee by chiral analytical SFC. Anal. Calcd for C19H28N6O4-H2O: C, 54.02; H, 7.16; N, 19.89. Found: C, 53.94; H, 7.22; N, 19.81.
PATENT
WO2020157652 EX 13
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020157652&_cid=P22-MDFD2U-50269-1
Example 13: (1R,3S)-3-[3-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}-amino)-1H-pyrazol-5-yl]cyclopentyl propan-2-ylcarbamate
(1R,3S)-3-[3-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-5-yl]cyclopentyl propan-2-ylcarbamate (Example 13, 2.06 g, 78%) was obtained as a white crystalline solid found to be a monohydrate (Form 1) based on elemental analysis. MS: 405 [M+H]+.1H NMR (400MHz, DMSO-d6) d = 12.23 (br s, 1H), 10.73 (br s, 1H), 7.11 (s, 1H), 6.96 (br d, J=7.0 Hz, 1H), 6.41 (br s, 1H), 5.00 (br s, 1H), 4.33 (s, 2H), 4.04 (s, 3H), 3.57 (qd, J=6.6, 13.4 Hz, 1H), 3.26 (s, 3H), 3.17-2.96 (m, 1H), 2.48-2.39 (m, 1H), 2.03 (br d, J=6.8 Hz, 1H), 1.95-1.83 (m, 1H), 1.73 (br d, J=8.5 Hz, 2H), 1.61 (br s, 1H), 1.03 (br d, J=6.3 Hz, 6H). Optical rotation [a]D +4.8 (c 1.0, MeOH). Chiral purity: >99% ee by chiral analytical SFC. Anal. Calcd for C19H28N6O4-H2O: C, 54.02; H, 7.16; N, 19.89. Found: C, 53.94; H, 7.22; N, 19.81.
The white crystalline solid from above (500 mg) was recrystallized from 9: 1 H2O/CH3CN (2 mL) by heating until dissolved and then allowing the resulting solution to stand at room temperature for 18 h. During the 18 h time period, larger crystals of monohydrate (Form 1) formed. Single crystal X-ray diffraction of a selected crystal from this material provided the structure in FIG.1.
PATENTS
WO2022018667
WO2022174031
WO2022137106
[1]. Douglas Carl BEHENNA, et al. Cdk2 inhibitors. WO2020157652A2.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
///////////Tagtociclib, PF-07104091, 2460249-19-6, Tegtociclib, XBD0JF5EHJ, PF 07104091