














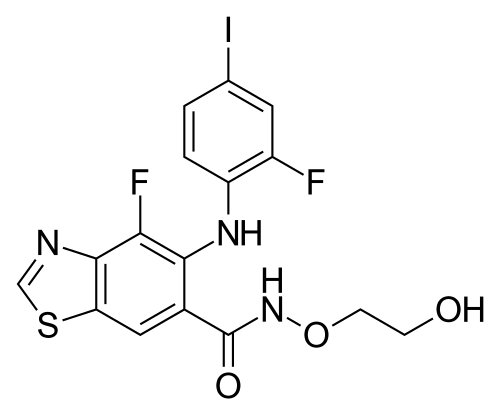
Tunlametinib
- CAS 1801756-06-8
- IF25NR1PV3
- HL085
- C16H12F2IN3O3S
491.3 g/mol
4-fluoro-5-(2-fluoro-4-iodoanilino)-N-(2-hydroxyethoxy)-1,3-benzothiazole-6-carboxamide
- 4-Fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-hydroxyethoxy)-6-benzothiazolecarboxamide
- 4-fluoro-5-(2-fluoro-4-iodoanilino)-N-(2-hydroxyethoxy)-1,3-benzothiazole-6-carboxamide
- 6-Benzothiazolecarboxamide, 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-hydroxyethoxy)-
Tunlametinib, an oral selective inhibitor of mitogen-activated protein kinase kinase 1 and 2 (MEK1/2), was developed by Shanghai KeChow Pharmaceuticals Co., Ltd. Marketed under the brand name
Keluping,
Tunlametinib is a pharmaceutical drug for the treatment of cancer. It is an inhbitor of mitogen-activated protein kinase kinase.[1]
In China, tunlametinib was approved in 2024 for the treatment of patients with NRAS-mutated advanced melanoma who were previously treated with a PD-1/PD-L1 targeting agent.[2][3]
It is also being studied for use in combination with vemurafenib in patients with advanced BRAF V600-mutant solid tumors.[4]
PAT
US9937158
PAT
https://patents.google.com/patent/WO2013107283A1/en
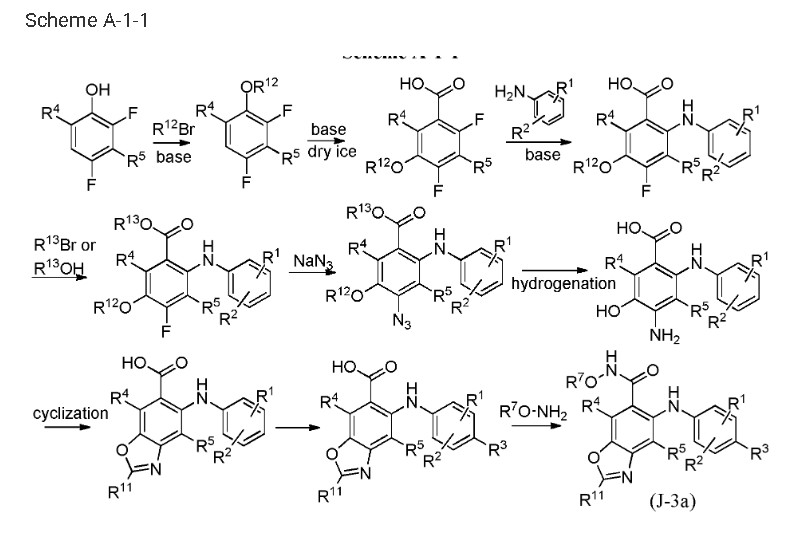
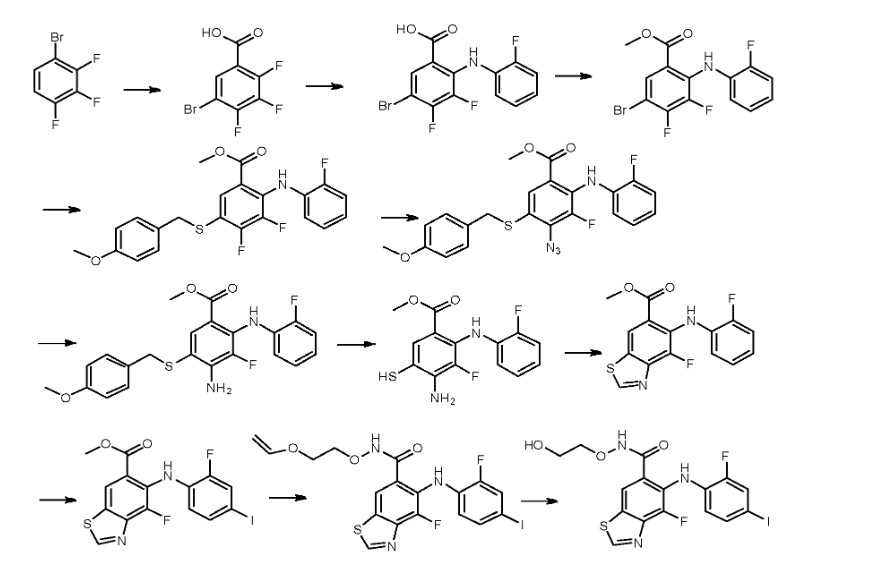
Step 1:

[0435] To a solution of 2,3,4-trifluorobromobenzene in appropriate solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), sulfolane, HMPA, DMPU, prefer anhydrous THF, ethyl ether and dioxane) was added strong base (such as LDA, nBuLi,
LiHDMS) at low temperature (-50 °C 80 °C, prefer -78 °C) under nitrogen atmosphere. The reaction is kept stirring for some time (0.5-12 h, prefer 0.5-2 h) and is added dry ice. After several hours (3-12 h, prefer 5-10 h), 5-bromo-2,3,4-trifluorobenzoic acid is obtained after conventional workup.
Step 2:

[0436] 5-Bromo-2,3,4-trifluorobenzoic acid can be reacted with halogenated aniline (such as o-fluoroaniline, o-chloroaniline, o-bromoaniline, o-iodoaniline) in the presence of base (such as LDA, n-BuLi, LiHDMS) in appropriate solvent (include aliphatic and aromatic
hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2- methoxyethyl ether, tetrahydrofuran, dioxane), sulfolane, HMPA, DMPU, prefer anhydrous THF, ethyl ether and dioxane) at low temperature (-50 °C— -80 °C, prefer -78 °C) for some time (such as 3-12 h, prefer 5-10 h). 5-Bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoic acid is obtained after conventional workup.
Step 3:

[0437] 5-Bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoic acid can be reacted with MeOH in the presence of SOCl2 in appropriate solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile(such as acetonitrile, propiononitrile), amide(such as N,N-dimethylformamide, N,N-dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer methanol and ethanol). The reaction proceeds for several hours (3-12 h, prefer 5-10 h). Methyl 5-bromo-3,4-difluoro-2-((2-fluorophenyl) amino)benzoate is obtained after conventional workup.
Step 4:

[0438] To a solution of methyl 5-bromo-3,4-difluoro-2-((2-fluorophenyl) amino)benzoate in appropriate solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ester(such as ethyl acetate, methyl acetate), nitrile(such as acetonitrile, propiononitrile), amide(such as N,N-dimethylformamide, N,N-dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer dioxane) was added base (such as aliphatic and aromatic amine(such as, but not limited to, N-ethyl-N-isopropylpropan-2-amine, triethylamine, diethylamine, DBU, t-butylamine, cyclopropanamine, dibutylamine, diisopropylamine, 1,2- dimethylpropanamine), inorganic base(such as Na2C03, K2C03, NaHC03, KHC03, t-BuONa, t- BuOK), prefer N-ethyl-N-isopropylpropan-2-amine) at ambient temperature under nitrogen atmosphere, followed by Pd catalyst (such as tris(dibenzylideneacetone)dipalladium,
bis(dibenzylideneacetone) palladium, bis(triphenylphosphine)palladium(II) chloride, palladium diacetate, tetrakis(triphenylphosphine)palladium, bis(triphenylphosphinepalladium)acetate, prefer tris(dibenzylideneacetone) dipalladium) and phosphine ligand (such as
dimethylbisdiphenylphosphinoxanthene, tri-tert-butylphosphine, tri-p-tolylphosphine, tris(4- chlorophenyl)phosphine, triisopropylphosphine, tris(2,6-dimethoxyphenyl)phosphine, 1, 1 ‘- bis(diphenylphosphino)ferrocene, prefer dimethylbisdiphenylphosphinoxanthene). The reaction is kept stirring at high temperature (80-130 °C, prefer 90-110 °C) for some time (8-24 h, prefer 12-18 h). Methyl 3,4-difluoro-2- ((2-fluorophenyl)amino)-5-((4-methoxybenzyl)thio)benzoate is obtained after conventional workup. Step 5:

[0439] Methyl 3,4-difluoro-2-((2-fluorophenyl)amino)-5-((4-methoxy benzyl)thio)benzoate can be reacted with azide (such as NaN3, KN3) at high temperature (60-120 °C, prefer 80-100 °C) in appropriate solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile (such as acetonitrile, propiononitrile), amide (such as N,N-dimethylformamide, N,N-dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer N,N-dimethylformamide and N,N-dimethylacetamide) for some time (1-12 h, prefer 3-10 h). Methyl 4-azido-3-fluoro-2-((2-fluorophenyl) amino)-5-((4-methoxybenzyl)thio)benzoate is obtained after conventional workup.
Step 6:

[0440] Methyl 4-azido-3-fluoro-2-((2-fluorophenyl)amino)-5-((4-methoxy
benzyl)thio)benzoate can be hydrogenated catalyzed by appropriate catalyst (such as Pd/C, Pt, Ni) in the solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ester(such as ethyl acetate, methyl acetate), amide (such as N,N-dimethylformamide, N,N- dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer methanol, ethanol, propan-l-ol and water) for some time (1-12 h, prefer 3-10 h). Methyl 4- amino-3-fluoro-2-((2-fluorophenyl)amino)-5-((4-methoxybenzyl)thio)benzoate is obtained after conventional workup. Step 7:

[0441] 4-Amino-3-fluoro-2-((2-fluorophenyl)amino)-5-((4-methoxybenzyl)thio)benzoate can be deprotected in the presence of acid (such as CF3COOH, HCOOH, CH3COOH and n- C5H11COOH, prefer CF3COOH) at certain temperature (20-75 °C, prefer 25-75 °C) in
appropriate aromatic aliphatic ether (such as anisole and phenetole, prefer anisole) for some time (1-12 h, prefer 3-10 h). Methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5- mercaptobenzoate is obtained after conventional workup.
Step 8:

[0442] Methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5-mercapto benzoate can be cyclized in the presence of acid (such as ^-toluenesulfonic acid, pyridinium toluene-4- sulphonate, formic acid, acetic acid, sulfuric acid) in appropriate solvent (include aliphatic and aromatic hydrocarbon (such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as
dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile (such as acetonitrile, propiononitrile), amide (such as N,N-dimethylformamide, N,N-dimethylacetamide and N- methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer methyl acetate, ethyl acetate and trimethoxymethane) for some time (0.2-12 h, prefer 0.5-10 h). Methyl 4-fluoro-5-((2- fluorophenyl)amino) benzo[d]thiazole-6-carboxylate is obtained after conventional workup. Step 9:

[0443] Methyl 4-fluoro-5-((2-fluorophenyl)amino)benzo[d]thiazole-6- carboxylate can be reacted with halogenations reagent (such as NIS) in the presence of acid (such as trifluoroacetic acid, trifluoromethanesulfonic acid, methanesulfonic acid, formic acid, acetic acid) at ambient temperature in appropriate solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile (such as acetonitrile, propiononitrile), amide (such as N,N- dimethylformamide, N,N-dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer N,N-dimethylformamide and N,N-dimethylacetamide) for some time (1- 12 h, prefer 3-10 h). Methyl 4-fluoro-5-((2-fluoro-4-iodophenyl) amino)benzo[d]thiazole-6- carboxylate is obtained after conventional workup.
Step 10:

[0444] 4-Fluoro-5-((2-fluoro-4-iodophenyl)amino)benzo[d]thiazole-6-carboxylic acid can be reacted with O-(2-(vinyloxy)ethyl)hydroxylamine in the presence of coupling reagent(such as HOBt, EDCI, HATU, TBTU) at ambient temperature in appropriate solvent(include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile (such as acetonitrile, propiononitrile), amide (such as N,N-dimethylformamide, N,N-dimethylacetamide and N- methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer dichloromethane, 1,2- dichloroethane and N,N-dimethylformamide) for some time (1-12 h, prefer 3-10 h). 4-Fluoro-5- ((2-fluoro-4-iodophenyl) amino)-N-(2-(vinyloxy)ethoxy)benzo[d]thiazole-6-carboxamide is obtained after conventional workup. Step 11:

[0445] 4-Fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-(vinyloxy)ethoxy)benzo[d]thiazole- 6-carboxamide can be reacted in the presence of acid (such as HCl, H2S04, trifluoroacetic acid) in appropriate solvent (include aliphatic and aromatic hydrocarbon (such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile (such as acetonitrile, propiononitrile), amide (such as N,N-dimethylformamide, N,N-dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer dichloromethane and 1,2-dichloroethane) for some time (1-12 h, prefer 3-10 h). 4-Fluoro- 5-((2-fluoro-4-iodophenyl)amino)-N-(2-hydroxy ethoxy)benzo[d]oxazole-6-carboxamide is obtained after conventional workup.
Example 9: Preparation of 4-fluoro-5-((2-fluoro-4-iodophenyDamino)-N-(2- hydroxyethoxy)benzo[d]thiazole-6-carboxamide (Compound 9)

Step 1: 5-bromo-2,3,4-trifluorobenzoic acid
[0510] To a solution of diisopropylamine (10.14 g, 100.20 mmol) in THF (100 mL) was added «-BuLi (40.08 mL, 2.5 M in hexane, 100.20 mmol) at -78 °C under nitrogen atmosphere. The stirring was maintained at this temperature for 1 h. Then a solution of l-bromo-2,3,4- trifluorobenzene (17.62 g, 83.50 mmol) in THF (120 mL) was added. After stirring for 1 h at -78 °C, the mixture was transferred to a bottle with dry ice. The mixture was stirred overnight at room temperature. The reaction was quenched with 10% aqueous HCl and pH was adjusted to 1- 2. The mixture was extracted with ethyl acetate (100 mL x 3). The combined organic extracts were washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated under reduced pressure to afford the desired product (20.12 g, 94.5% yield). 1H NMR (400 MHz, DMSO-d6): δ 13.95 (s, 1H), 7.97 (m, 1H).
Step 2: 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoic acid
[0511] To a solution of 2-fluoroaniline (17.54 g, 157.80 mmol) and 5-bromo-2,3,4- trifluorobenzoic acid (20.12 g, 78.90 mmol) in THF (120 mL) was added LiHMDS (236.7 mL, 1 M in THF, 236.7 mmol) dropwisely at -78 °C under nitrogen atmosphere. The mixture was allowed to slowly warm to room temperature and stirred at this temperature overnight. The reaction was quenched with water (100 mL) and acidified to pH 2-3 with 10% HCl (aq.). The mixture was extracted with ethyl acetate (100 mL χ 3). The combined organic extracts were washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo to afford the desired product (pale yellow solid, 24.24 g, 88.8% yield). 1H NMR (400 MHz, DMSO-d6): δ 9.22 (s, 1H), 8.01 (dd, J= 7.4, 2.1 Hz, 1H), 7.25 (m, 1H), 7.10 (m, 3H).
Step 3: methyl 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoate
[0512] To a solution of 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino) benzoic acid (24.24 g, 70.04 mmol) in MeOH (300 mL) was added thionyl chloride (20 mL). After stirring at 85 °C overnight, most MeOH was removed in vacuo. The residue was neutralized with saturated sodium bicarbonate (aq.) and extracted with ethyl acetate (100 mL χ 3). The combined organic layer was washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated. After purification by column chromatography on silica gel (petroleum ether/ethyl acetate, 50: 1, v/v), the corresponding product was obtained as a white solid (22.33 g, 88.5% yield). 1H NMR (400 MHz, CDC13): δ 9.06 (s, 1H), 8.01 (dd, J= 7.1, 2.3 Hz, 1H), 7.04 (m, 4H), 3.92 (s, 3H).
Step 4: methyl 3,4-difluoro-2-((2-fluorophenyl)amino)-5-((4-methoxybenzyl)thio)benzoate
[0513] To a solution of methyl 5-bromo-3,4-difluoro-2-((2-fluorophenyl) amino)benzoate (22.33 g, 62.01 mmol) in anhydrous 1,4-dioxane (200 mL) was added N,N- diisopropylethylamine (16.03 g, 124.04 mmol). Then Pd2(dba)3 (2.84 g, 3.10 mmol) followed by Xantphos (3.59 g, 6.20 mmol) and 4-methoxy-a-toluenethiol (10.27 g, 65.11 mmol) was added under nitrogen atmosphere. The mixture was stirred overnight at 100 °C under N2 atmosphere and then allowed to warm to ambient temperature. The insoluble matter was filtered off and the filter cake was washed ethyl acetate. The filtrate was diluted with water (300 mL) and extracted with ethyl acetate (100 mL x 3). The combined organic layers were washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated. The crude product was purified by column chromatography on silica gel (petroleum ether/ethyl acetate, 50: 1, v/v) to give the desired product (pale yellow solid, 24.35 g, 90.6% yield). 1H NMR (400 MHz, CDC13): δ 9.12 (s, 1H), 7.78 (d, 1H), 7.25 (m, 6H), 6.85 (m, 2H), 4.03 (s, 2H), 3.90 (s, 3H), 3.80 (s, 3H). Step 5: methyl 4-azido-5-(4-methoxybenzylthio)-3-fluoro-2-((2-fluorophenyl)amino)benzoate
[0514] To a solution of methyl 5-(4-methoxybenzylthio)-3,4-difluoro-2- ((2- fluorophenyl)amino)benzoate (24.35 g, 56.18 mmol) in DMF (200 mL) was added NaN3 (4.38 g, 67.41 mmol) at ambient temperature. The mixture was stirred at 90 °C for 3 h. Then water (200 mL) was added. The solution was extracted with ethyl acetate (100 mL χ 3). The combined organic extracts were washed with water (100 mL) and brine (100 mL), dried over Na2S04 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate, 10: 1, v/v) and gave the desired product (white solid, 21.04 g, 82.1% yield). 1H NMR (400 MHz, CDC13): δ 8.98 (s, 1H), 7.75 (s, 1H), 7.10 (m, 6H), 6.84 (m, 2H), 4.03 (s, 2H), 3.92 (s, 3H), 3.81 (s, 3H). Step 6: methyl 4-amino-5-(4-methoxybenzylthio)-3-fluoro-2-((2-fluorophenyl)amino)benzoate To a solution of methyl 4-azido-5-(4-methoxybenzylthio)-3-fluoro-2-((2- fluorophenyl)amino)benzoate (21.04 g, 46.09 mmol) in MeOH (500 mL) was added and 10% palladium on carbon (3.40 g) under nitrogen atmosphere. Then the nitrogen atmosphere was completely changed to hydrogen atmosphere. The mixture was stirred for 2 h at ambient temperature. After the insoluble matter was filtered off, the solvent was evaporated in vacuo to give the desired product (19.46 g, 98.1% yield). 1H NMR (400 MHz, CDC13): δ 9.07 (s, 1H), 7.77 (s, 1H), 7.06 (m, 4H), 6.95 (m, 2H), 6.81 (d, J = 8.3 Hz, 2H), 4.68 (s, 2H), 3.85 (s, 5H), 3.81 (s, 3H).
Step 7: dimethyl 5,5′-disulfanediylbis(4-amino-3-fluoro-2-((2-fluorophenyl)amino)benzoate)
[0515] To a solution of methyl 4-amino-5-(4-methoxybenzylthio)-3-fluoro-2-((2- fluorophenyl)amino)benzoate (19.46 g, 45.21 mmol) in CH2C12 (180 mL) was added DDQ (11.29 g, 49.73 mmol) followed by water (20 mL). After stirring at ambient temperature for 10 h, the reaction was quenched by saturated sodium bicarbonate (aq., 100 mL). The aqueous layer was extracted by CH2C12 (100 mL χ 3). The combined organic phase was washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated. The crude product was purified by column chromatography on silica gel (petroleum ether/ethyl acetate, 5: 1, v/v) to give the desired product (pale yellow solid, 9.81 g, 35.1% yield). 1H NMR (400 MHz, CDC13): δ 9.34 (s, 2H), 7.46 (s, 2H), 7.06 (m, 8H), 4.89 (br, 4H), 3.75 (s, 6H). Step 8: methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5-mercaptobenzoate
[0516] To a solution of dimethyl 5,5′-disulfanediylbis(4-amino-3-fluoro-2-((2- fluorophenyl)amino)benzoate) (9.81 g, 15.86 mmol) in THF/MeOH (100 mL, 10: 1, v/v) was added NaBH4 (3.00 g, 79.29 mmol) portion-wise in 1 h. After stirring at ambient temperature for 1 h, the reaction was quenched with 10% HCl (aq.) and pH was adjusted to 1-2. The aqueous layer was extracted with CH2C12 (50 mL χ 3). The combined organic phase was washed with water (50 mL) and brine (50 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo. The crude product was used directly in the next step without further purification.
Step 9: methyl 4-fluoro-5-((2-fluorophenyl)amino)benzofdJthiazole-6-carboxylate
[0517] To a solution of methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5- mercaptobenzoate in trimethyl orthoformate (50 mL) was added p-TsOU (0.61 g, 3.17 mmol). The reaction mixture was stirred for 1 h and treated with water (100 mL). The precipitate was filtered off and the filter cake was washed with water to afford the desired product (pale yellow solid, 8.64 g, 85.1% yield for two steps). 1H MR (400 MHz, CDC13): δ 9.13 (s, 1H), 8.68 (s, 1H), 8.46 (s, 1H), 7.10 (m, 1H), 7.01 (m, 1H), 6.92 (s, 2H), 3.97 (s, 3H).
Step 10: methyl 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)benzofdJthiazole-6-carboxylate
[0518] To a solution of methyl 4-fluoro-5-((2-fluorophenyl)amino)benzo[d]thiazole-6- carboxylate (8.64 g, 26.97 mmol) in DMF (100 mL) was added NIS (6.68 g, 29.67 mmol) followed by trifluoroacetic acid (0.5 mL). After stirring for 5 h at ambient temperature, the reaction was treated by water (150 mL). The precipitate was filtered off and the filter cake was washed with water. The desired product was obtained as a yellow solid (10.34 g, 86.0% yield). 1H NMR (400 MHz, CDC13): δ 9.14 (s, 1H), 8.66 (s, 1H), 8.46 (s, 1H), 7.42 (d, J= 10.4 Hz, 1H), 7.31 (d, J= 8.8 Hz, 1H), 6.63 (dd, J= 15.0, 8.7 Hz, 1H), 3.97 (s, 3H).
Step 11: 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)benzo[d]thiazole-6-carboxylic acid
[0519] To a solution of methyl 4-fluoro-5-((2-fluoro-4-iodophenyl)amino) benzo[d]thiazole-6- carboxylate (10.34 g, 23.17 mmol) in THF and MeOH (20 mL, 4: 1, v/v) was added 5.0 M LiOH (aq., 2 mL, 10 mmol). After stirring at ambient temperature for 2 h, the reaction was treated with 1.0 M HCl (aq.) till the solution was acidic. The aqueous layer was extracted with ethyl acetate (50 mL x 3). The combined organic phase was washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated to give the desired product (9.51 g, 95.0% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.10 (s, 1H), 9.18 (s, 1H), 8.68 (s, 1H), 8.45 (s, 1H), 7.41 (m, 1H), 7.30 (m, 1H), 6.65 (m, 1H). Step 12: 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-(vinyloxy)etho
carboxamide
[0520] To a solution of 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)benzo[d]thiazole-6- carboxylic acid (519 mg, 1.20 mmol) in CH2C12 (10 mL) was added HOBt (254 mg, 1.63 mmol) and EDCI (314 mg, 1.63 mmol). The mixture was stirred for 1 h and O-(2-
(vinyloxy)ethyl)hydroxyl -amine (172 mg, 1.62 mmol) was added. After stirring for 4 h at ambient temperature, the reaction was treated with saturated H4C1 (aq.). The resultant mixture was extracted with CH2C12 (30 mL χ 3). The combined organic extracts were washed with water (30 mL) and brine (30 mL), dried over Na2S04 filtered, and concentrated in vacuo. The crude product (492 mg) was used directly in the next step without further purification.
Step 13: 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-hydroxyethoxy)benzo[d]thiazole-6- carboxamide
[0521] To a solution of 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2- (vinyloxy)ethoxy)benzo[d]thiazole-6-carboxamide (492 mg, 1.00 mmol) in CH2C12 (10 mL) was added 1.0 N HCl (aq., 5 mL, 5 mmol). After stirring for 1 h, the reaction mixture was neutralized with saturated NaHC03 (aq.). The aqueous layer was washed with CH2C12 (30 mL). The combined organic layer was washed with water (30 mL x 2) and brine (30 mL), dried over Na2S04, filtered and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (CH2Cl2/MeOH, 50: 1, v/v) and gave the desired product as a white solid (446 mg, 75.9% yield for the two steps). 1H MR (400 MHz, DMSO-d6): δ 11.80 (s, 1H), 9.55 (s, 1H), 8.22 (s, 1H), 8.12 (s, 1H), 7.55 (d, J= 11.0 Hz, 1H), 7.31 (d, J= 8.5 Hz, 1H), 6.48 (d, J= 9.2 Hz, 1H), 4.72 (s, 1H), 3.84 (m, 2H), 3.57 (m, 2H). MS APCI(+)m/z: 491.8, [M+H].
Example 9A: Preparation of 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2- hydroxyethoxy)benzo[d]thiazole-6-carboxamide (Compound 9)

Step 1: 5-bromo-2,3,4-trifluorobenzoic aci

[0522] To a solution of l-bromo-2,3,4-trifluorobenzene (13.64 g, 64.6 mmol) in THF (120 mL) was added lithium diisopropylamide (2.0 M in THF, 33.9 mL, 67.8 mmol) at -78 °C under nitrogen atmosphere. After stirring for 1 h at -78 °C, the mixture was transferred to a bottle with dry ice. The mixture was stirred overnight at room temperature. The reaction was quenched with 10% aqueous HC1 (300 mL) and extracted with ethyl acetate (200 mL x 3). The combined organic extracts were washed with 5% sodium hydroxide (300 mL). The aqueous layer was acidized to pH 1 and extracted with ethyl acetate (200 mL χ 3). The combined organic extract was dried over Na2S04, filtered and concentrated under reduced pressure to afford the desired product (white solid, 13.51 g, 82% yield). 1H MR (400 MHz, CDC13): δ 13.94 (s, 1H), 7.95 (m,
1H).
Step 2: 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoic

[0523] To a solution of 2-fluoroaniline (10.2 mL, 105.8 mmol) and 5-bromo-2,3,4- trifluorobenzoic acid (13.51 g, 52.9 mmol) in THF (120 mL) was added LiHMDS (158.7 mL, 1 M in THF, 158.7 mmol) dropwisely at -78 °C under nitrogen atmosphere. The mixture was allowed to slowly warm to room temperature and stirred at this temperature overnight. The reaction was quenched with 10% HC1 (aq., 100 mL) and extracted with ethyl acetate (200 mL x 3). The combined organic extracts were washed with water (200 mL x 3) and brine (200 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo to afford the desired product (pale yellow solid, 13.73 g, 75% yield). 1H MR (400 MHz, DMSO-d6): δ 9.21 (s, 1H), 8.01 (d, 1H), 7.26 (m, 1H), 7.01-7.16 (m, 3H).
Step 3: methyl 5-bromo-3,4-difluoro-2- -fluorophenyl)amino)benzoate

[0524] To a solution of 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoic acid (13.73 g, 39.6 mmol) in MeOH (300 mL) was added SOCl2 (60 mL). After stirring at 85 °C overnight, most MeOH was removed in vacuo. The residue was neutralized with saturated sodium bicarbonate (aq.) and extracted with ethyl acetate (300 mL χ 3). The combined organic extract was washed with water (200 mL x 3) and brine (200 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo to afford the corresponding product (gray solid, 12.58 g, 90% yield). 1H MR (400 MHz, CDC13): δ 9.09 (s, 1H), 8.05 (d, 1H), 7.00-7.14 (m, 4H), 3.94 (s, 3H).
Step 4: methyl 3,4-difluoro-2-((2-fluorophenyl)amino)-5-((4-methoxybenzyl)thio)benzoate

[0525] To a solution of methyl 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoate (12.85 g, 35.6 mmol) in anhydrous 1,4-dioxane (30 mL) was added N,N-diisopropylethylamine (9.21 g, 71.2 mmol). Then Pd2(dba)3 (1.63 g, 1.78 mmol) followed by Xantphos (2.06 g, 3.56 mmol) and 4-methoxy-a-toluenethiol (5.48 g, 35.6 mmol) was added under nitrogen atmosphere. The mixture was stirred overnight at 100 °C under N2 atmosphere and then allowed to cool to ambient temperature. The reaction was quenched with water (150 mL) and extracted with ethyl acetate (200 mL χ 3). The combined organic extract was washed with water (200 mL χ 3) and brine (200 mL) sequentially, dried over Na2S04, filtered and concentrated. The crude product was purified by column chromatography on silica gel (petroleum ether/ethyl acetate, 50: 1, v/v) to give the desired product (pale yellow solid, 12.64 g, 82% yield). 1H NMR (400 MHz, CDC13): δ 9.12 (s, 1H), 7.78 (d, 1H), 7.06-7.44 (m, 6H), 6.82-6.88 (m, 2H), 4.03 (s, 2H), 3.90 (s, 3H), 3.80 (s, 3H).
Step 5: methyl 4-azido-5-(4-methoxybenzylthio)-3-fluoro-2-((2-fluorophenyl)amino)benzoate

[0526] To a solution of methyl 5-(4-methoxybenzylthio)-3,4-difluoro-2-((2- fluorophenyl)amino)benzoate (12.64 g, 29.2 mmol) in DMF (30 mL) was added NaN3 (2.28 g, 35.0 mmol) at ambient temperature. The mixture was stirred at 90 °C for 3 h. Then water (150 mL) was added. The solution was extracted with ethyl acetate (100 mL χ 3). The combined organic extracts were washed with water (100 mL χ 3) and brine (100 mL), dried over Na2S04 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate, 10: 1, v/v) and gave the desired product (white solid, 10.38 g, 78% yield). 1H NMR (400 MHz, CDC13): δ 8.98 (s, 1H), 7.75 (s, 1H), 7.02-7.28 (m, 6H), 6.83- 6.85 (m, 2H), 4.03 (s, 2H), 3.92 (s, 3H), 3.81 (s, 3H).
Step 6: methyl 4-amino-5-(4-methoxybenzylthio)-3-fluoro-2-((2-fluorophenyl)amino)benzoate

[0527] To a solution of methyl 4-azido-5-(4-methoxybenzylthio)-3-fluoro-2-((2- fluorophenyl)amino)benzoate (10.38 g, 22.7 mmol) in MeOH (100 mL) was added and 10% palladium on carbon (1.55 g) under nitrogen atmosphere. Then the nitrogen atmosphere was completely changed to hydrogen atmosphere. The mixture was stirred at ambient temperature for 6 h. After the insoluble matter was filtered off, the solvent was evaporated in vacuo to give the desired product (9.79 g, 100% yield).1H MR (400 MHz, CDC13): δ 9.08 (s, 1H), 7.78 (s, 1H), 6.93-7.28 (m, 8H), 4.65 (s, 2H), 4.00 (s, 2H), 3.89 (s, 3H), 3.75 (s, 3H).
Step 7: methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5-mercaptobenzoate

[0528] To a solution of methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5-((4- methoxybenzyl)thio)benzoate (9.79 g, 22.7 mmol) in anisole (12 mL) was added CF3COOH (20 mL). After stirring at ambient temperature for 23 h, the solvent was removed in vacuo. To the residue was added water (30 mL). The mixture was neutralized with 25% aqueous ammonia and extracted with ethyl acetate (100 mL χ 3). The combined organic layer was washed with water (100 mL x 3) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated to give the desired product (white solid, 5.28 g, 75% yield). The product was used directly in the next step without further purification.
Step 8: methyl 4-fluoro-5-((2-fluorophenyl)amino)benzofdJthiazole-6-carboxylate

[0529] To a solution of methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5- mercaptobenzoate (2.07 g, 6.67 mmol) in trimethyl orthoformate (20 mL) was added p-TsOU (166 mg, 0.65 mmol). The reaction mixture was stirred for 1 h and treated with water (100 mL). The precipitate was filtered off and the filter cake was washed with water to afford the desired product (white solid, 1.963 g, 92% yield for two steps). 1H NMR (400 MHz, DMSO-d6): δ 9.01 (s, 1H), 8.08 (s, 1H), 7.90 (s, 1H), 7.15-6.78 (m, 4H), 3.91 (s, 3H).
Step 9: methyl 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)benzofdJthiazole-6-carboxylate

[0530] To a solution of methyl 4-fluoro-5-((2-fluorophenyl)amino)benzo[d]thiazole-6- carboxylate (1.963 g, 6.14 mmol) in DMF (10 mL) was added NIS (1.5 g, 6.5 mmol) followed by trifluoroacetic acid (0.5 mL). After stirring for 4 h at ambient temperature, the reaction was treated by saturated H4C1 (aq.). The aqueous layer was extracted with ethyl acetate (150 mL χ 3). The combined organic layer was washed with water (100 mL x 3) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo. After purification by flash column chromatography on silica gel (petroleum ether/ethyl acetate, 10: 1, v/v), the desired product was obtained as white solid (1.889 g, 69% yield). 1H NMR (400 MHz, DMSO-d6): δ 9.03 (s, 1H), 8.10 (s, 1H), 7.93 (s, 1H), 7.18-6.72 (m, 3H), 3.91 (s, 3H).
Step 10: 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-(vinyloxy
carboxamide

[0531] To a solution of O-(2-(vinyloxy)ethyl)hydroxyl-amine (172 mg, 1.62 mmol) in THF (6 mL) was added LiHMDS (2.5 mL, 1 M in THF, 2.5 mmol) at -78 °C. After stirring at this temperature for 10 min, a solution of methyl 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)benzo[d] thiazole-6-carboxylate (360 mg, 0.81 mmol) in THF was syringed dropwisely. Then the mixture was allowed to warm to ambient temperature, quenched with saturated NH4C1 (aq., 20 mL) and extracted with ethyl acetate (15 mL χ 3). The combined organic extract was washed with water (10 mL x 3) and brine (10 mL), dried over Na2S04, filtered and concentrated in vacuo. After purification by flash chromatography (petroleum ether/ethyl acetate, 10: 1, v/v), the desired product was obtained (410 mg, 98% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.85 (s, 1H),
8.98 (s, 1H), 8.04 (s, 1H), 7.89 (s, 1H), 7.55 (d, J= 10.8 Hz, 1H), 7.31 (d, J = 8.1 Hz, 1H), 6.53 (dd, J= 13.9, 6.6 Hz, 1H), 6.42 (d, J= 6.0 Hz, 1H), 4.21 (d, J= 14.5 Hz, 1H), 4.01 (m, 3H), 3.83 (m, 2H).
Step 11: 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-hydroxyethoxy)benzofdJthiazole-6- carboxamide

[0532] To a solution of 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2- (vinyloxy)ethoxy)benzo[d]thiazole-6-carboxamide (410 mg, 0.8 mmol) in CH2C12 (5 mL) was added 1.0 N HCl (aq., 5 mL, 5 mmol) dropwise. After stirring for 1 h, the reaction mixture was neutralized with saturated NaHC03 (aq.). The organic layer was separated, washed with water (30 mL x 2) and brine (30 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (CH2Cl2/MeOH, 15: 1, v/v) and the desired product was obtained as a white solid (290 mg, 52 % yield). 1H MR (400 MHz, DMSO-de): δ 11.83 (s, 1H), 8.92 (s, 1H), 8.03 (s, 1H), 7.90 (s, 1H), 7.56 (d, J= 9.4 Hz, 1H), 7.30 (d, J= 8.7 Hz, 1H), 6.41 (m, 1H), 4.72 (m, 1H), 3.85 (m, 2H), 3.59 (m, 2H). MS (ES+): m/z 492.35 [MH+].
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Tunlametinib, an oral selective inhibitor of mitogen-activated protein kinase kinase 1 and 2 (MEK1/2), was developed by Shanghai KeChow Pharmaceuticals Co., Ltd. Marketed under the brand name
Keluping, it received conditional approval from the NMP in 2024 for the treatment of patients with advanced melanoma harboring NRAS mutations, particularly those who have not responded to anti-PD-1/PD-L1therapies [1]. Tunlametinib exerts its antitumor effects by targeting the MEK1/2 kinases within the RAS-RAF-MEK-ERK signaling pathway, thereby disrupting downstream signaling cascades and inhibiting tumor cell growth and proliferation [2]. Its clinical efficacy was demonstrated in a Phase II pivotal registration study (NCT05217303) involving patients with advanced NRAS-mutant melanoma [3]. The study reported a confirmed objective response rate (ORR) of 34.8 % and a median progression-free survival (mPFS) of 4.2 months. These findings suggest that Tunlametinib holds promise as a treatment option for NRAS-mutant melanoma, including in patients who have failed immunotherapy. In terms of safety, Tunlametinib has been generally well-tolerated [4]. Adverse events frequently encountered during treatment primarily consist of increased blood creatine phosphokinase (CPK) levels, diarrhea, and edema. Additionally, warnings and precautions pertinent to Tunlametinib therapy encompass decreased left ventricular ejection fraction (LVEF), skin toxicity, ocular toxicity, interstitial lung disease,
gastrointestinal reactions, and elevated CPK levels [5].
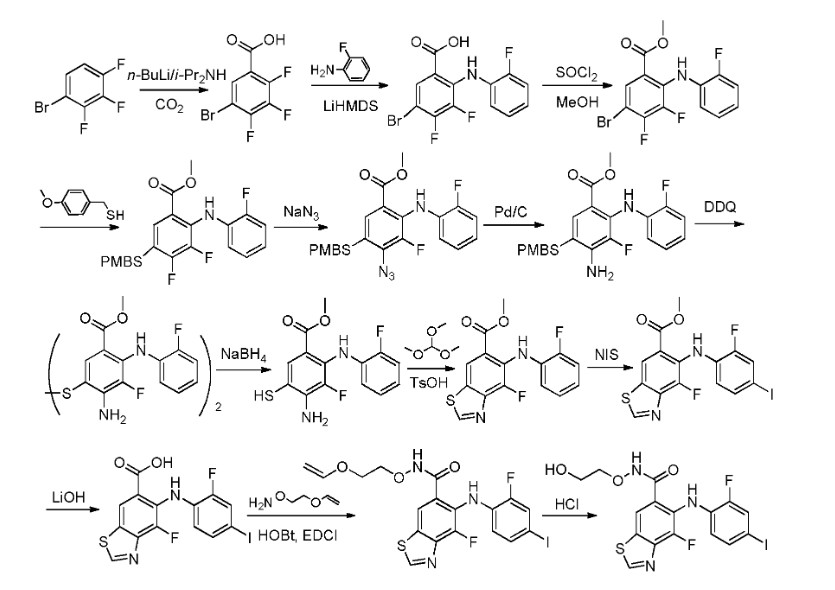
The synthetic pathway of Tunlametinib, illustrated in Scheme 1, begins with carboxylation of Tunl-001 to yield Tunl-002 [6]. Nucleophilic substitution of Tunl-002 with Tunl-003 then produces Tunl-004,
which undergoes esterification to form Tunl-005. Subsequent nucleophilic substitution between Tunl-05 and Tunl-006 generates Tunl-007. This intermediate undergoes azidation to afford Tunl-008, followed by
reduction to Tunl-009. Treatment of Tunl-009 with DDQ converts it to Tunl-010, which is deprotected to yield Tunl-011. Cycloaddition of Tunl-011 with Tunl-012 forms Tunl-013. Iodination of Tunl-013 gives
Tunl-014, which is hydrolyzed to produce Tunl-015. Amidation of Tunl-015 with Tunl-016 yields Tunl-017, and its subsequent acidolysis completes the synthesis of Tunlametinib.
[1] Y. Liu, Y. Cheng, G. Huang, X. Xia, X. Wang, H. Tian, Preclinical characterization of
tunlametinib, a novel, potent, and selective MEK inhibitor, Front. Pharmacol. 14
(2023) 1271268.
[2] S.J. Keam, Tunlametinib: first approval, Drugs 84 (2024) 1005–1010.
[3] X. Wei, Z. Zou, W. Zhang, M. Fang, X. Zhang, Z. Luo, J. Chen, G. Huang, P. Zhang,
Y. Cheng, J. Liu, J. Liu, J. Zhang, D. Wu, Y. Chen, X. Ma, H. Pan, R. Jiang, X. Liu,
X. Ren, H. Tian, Z. Jia, J. Guo, L. Si, A phase II study of efficacy and safety of the MEK inhibitor tunlametinib in patients with advanced NRAS-Mutant melanoma,
Eur. J. Cancer 202 (2024) 114008.
[4] Q. Zhao, T. Wang, H. Wang, C. Cui, W. Zhong, D. Fu, W. Xi, L. Si, J. Guo, Y. Cheng,
H. Tian, P. Hu, Phase I pharmacokinetic study of an oral, small-molecule MEK
inhibitor tunlametinib in patients with advanced NRAS mutant melanoma, Front.
Pharmacol. 13 (2022) 1039416.
[5] Y. Shi, X. Han, Q. Zhao, Y. Zheng, J. Chen, X. Yu, J. Fang, Y. Liu, D. Huang, T. Liu,
H. Shen, S. Luo, H. Yu, Y. Cao, X. Zhang, P. Hu, Tunlametinib (HL-085) plus
vemurafenib in patients with advanced BRAF V600-mutant solid tumors: an open-
label, single-arm, multicenter, phase I study, Exp. Hematol. Oncol. 13 (2024) 60.
[6] H. Tian, C. Ji, C. Liu, L. Kong, Y. Cheng, G. Huang, Benzoheterocyclic Compounds
and Use Thereof, 2014. US9937158B2.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- “Tunlametinib”. NCI Drug Dictionary. National Cancer Institute.
- “Tunlametinib Wins Approval in China for NRAS+ Advanced Melanoma After PD-1/PD-L1 Therapy”. 18 March 2024.
- Keam SJ (2024). “Tunlametinib: First Approval”. Drugs. 84 (8): 1005–1010. doi:10.1007/s40265-024-02072-x. PMID 39034326.
- Shi Y, Han X, Zhao Q, Zheng Y, Chen J, Yu X, et al. (2024). “Tunlametinib (HL-085) plus vemurafenib in patients with advanced BRAF V600-mutant solid tumors: An open-label, single-arm, multicenter, phase I study”. Experimental Hematology & Oncology. 13 (1): 60. doi:10.1186/s40164-024-00528-0. PMC 11167782. PMID 38867257.
| Clinical data | |
|---|---|
| Other names | HL-085 |
| ATC code | None |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1801756-06-8 |
| PubChem CID | 71621329 |
| ChemSpider | 115006753 |
| UNII | IF25NR1PV3 |
| ChEMBL | ChEMBL5095241 |
| Chemical and physical data | |
| Formula | C16H12F2IN3O3S |
| Molar mass | 491.25 g·mol−1 |
{kind=link}
/////////Tunlametinib, CHINA 2024, APPROVALS 2024, Shanghai KeChow, Keluping,1801756-06-8, IF25NR1PV3, HL 085