














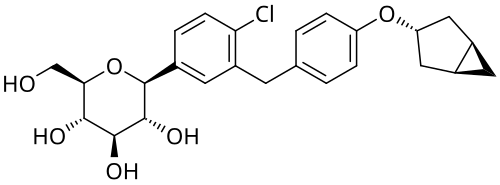
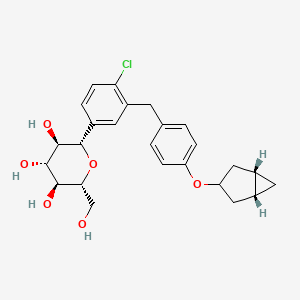
Janagliflozin
WeightAverage: 460.95
Monoisotopic: 460.1652664
Chemical FormulaC25H29ClO6
- WK4RT85HCA
- XZP-5695
- UNII-WK4RT85HCA
- 1800115-22-3
- (2S,3R,4R,5S,6R)-2-[3-[[4-[[(1S,5R)-3-bicyclo[3.1.0]hexanyl]oxy]phenyl]methyl]-4-chlorophenyl]-6-(hydroxymethyl)oxane-3,4,5-triol
- D-Glucitol, 1,5-anhydro-1-C-(3-((4-((1alpha,3alpha,5alpha)-bicyclo(3.1.0)hex-3-yloxy)phenyl)methyl)-4-chlorophenyl)-, (1S)-
- (2S,3R,4R,5S,6R)-2-[3-[[4-[[(1S,5R)-3-bicyclo[3.1.0]hexanyl]oxy]phenyl]methyl]-4-chlorophenyl]-6-(hydroxymethyl)oxane-3,4,5-triol
- D-GLUCITOL, 1,5-ANHYDRO-1-C-(3-((4-((1.ALPHA.,3.ALPHA.,5.ALPHA.)-BICYCLO(3.1.0)HEX-3-YLOXY)PHENYL)METHYL)-4-CHLOROPHENYL)-, (1S)-
- (2S,3R,4R,5S,6R)-2-(3-(4-(((1R,3s,5S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
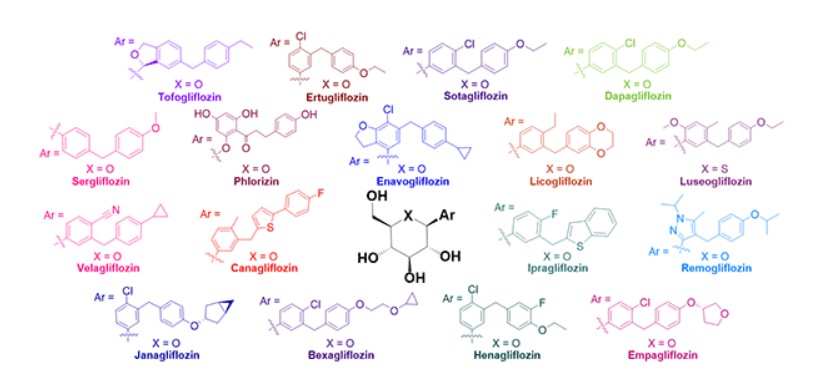
China 2024, approvals 2024, Jilin Huisheng Biopharmaceutical Co, sihuan, SGLT2 inhibitors, Huiyoujing
Janagliflozin is an SGLT2 inhibitor developed by Sihuan Pharmaceutical.[1][2][3][4][5][6] It is approved in China for the treatment of type 2 diabetes.[7]
PAPER
https://www.thieme-connect.de/products/ejournals/abstract/10.1055/s-0042-1751524
(71) (a) Wu, F. US9315438B2, 2016. (b) Wu, F. EP2891654A1, 2014.
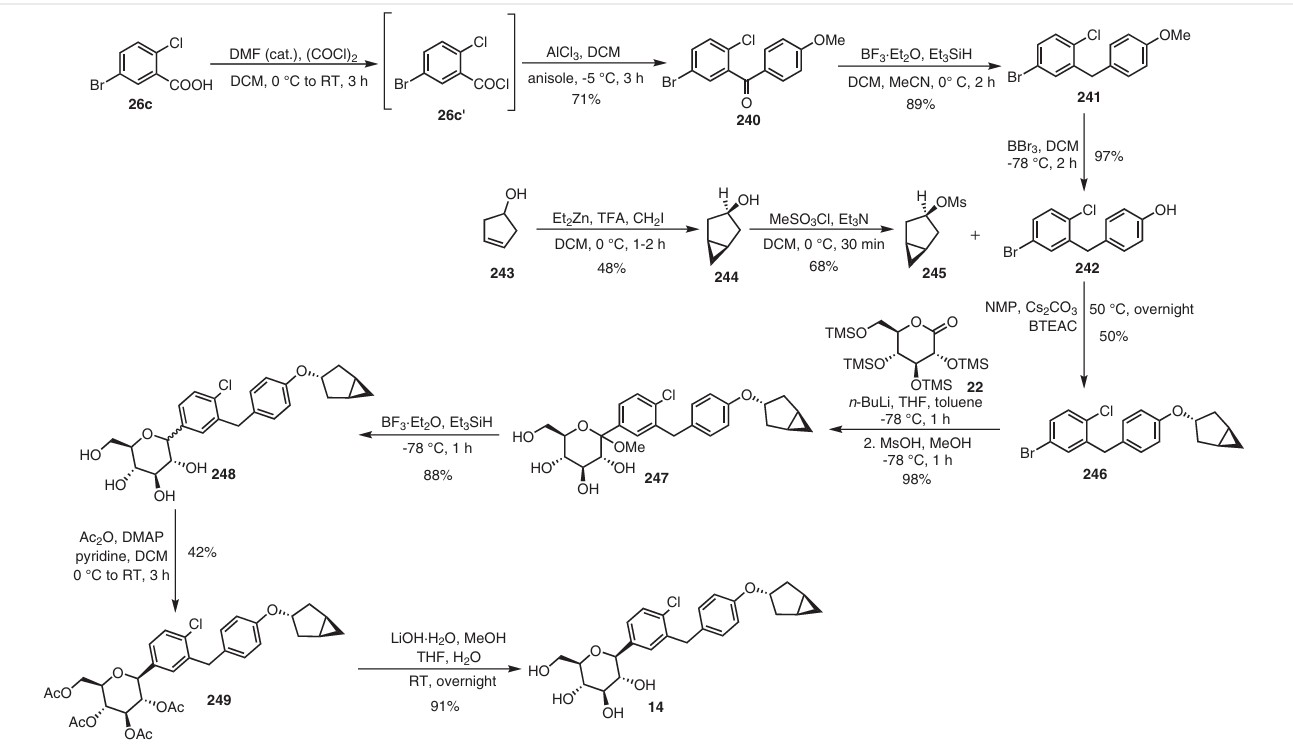
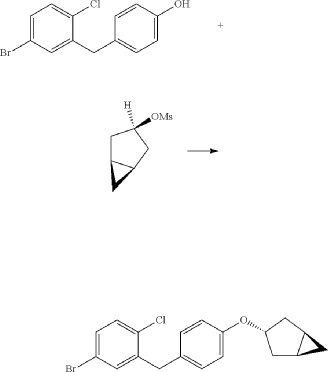
Initially, the two advanced intermediates were synthesized and then coupled under cryogenic conditions using nBuLi. The construction of 242 commences with the reaction of 5-bromo-2-chlorobenzoic acid (26c) with oxalyl chloride and a catalytic amount of DMF in DCM, yielding the acid chloride derivative 26c′. This intermediate is then subjected to Friedel–Crafts acylation with anisole to produce 240 in
71% yield. Subsequent reduction of 240 was carried out using boron trifluoride–diethyl etherate and triethylsilane in a DCM/acetonitrile mixture, leading to the formation of 241 in an excellent yield. Demethylation of compound 241 is accomplished using boron tribromide at low temperature, resulting in 242 with a yield of 97%. On the other hand, the synthesis of 245 involves two steps starting from commercially available cyclopent-3-en-1-ol (243). The Simmons Smith cyclopropanation of 243 is performed using a mixture of trifluoroacetic acid, diiodomethane, and diethylzinc in DCM, providing 244 with a yield of 48%. Compound 244 is then further treated with methanesulfonyl chloride to give the mesylated compound 245 in a yield of 68%. Subse quently, 4-(5-bromo-2-chlorobenzyl)phenol (242) is allowed to react with 245 in the presence of NMP, cesium carbonate, and BTEAC (benzyltriethylammonium chloride) to give 246. The next step involves a lithium–halogen exchange on
246 using n-butyllithium, with addition to 22 at –78 °C affording the hydroxy intermediate. Methylation of this hydroxy intermediate using methanesulfonic acid and methanol provides 247 in 98% yield. Reduction of 247 using borontrifluoride–diethyl etherate and triethylsilane at –78 °C furnishes 248. To achieve the desired isomer, all of the hydroxy groups of compound 248 were protected using acetic anhydride, DMAP, and pyridine in DCM at 0 °C to give the O-acylated compound 249. In the final step, 249 is hydrolyzed us ing lithium hydroxide monohydrate in a mixed solvent consisting of methanol, THF, and water to provide the desired compound janagliflozin (14) in a yield of 91%. This truncated synthetic route is protection-group-free, and is well suited for scale-up. The drawback of the synthetic route is
the late-stage enrichment of the desired isomer in the final product via acylated derivative 249. The poor isolated yield of 249 is not commercially favored due to low throughput and an increase in raw material and production costs.
PAPER
https://pubs.acs.org/doi/10.1021/acs.oprd.8b00017
SYN
https://www.sciencedirect.com/science/article/abs/pii/S022352342400223X
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US142552820&_cid=P11-MEPJES-88258-1
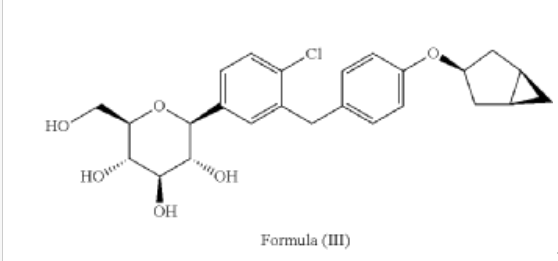
Example 1
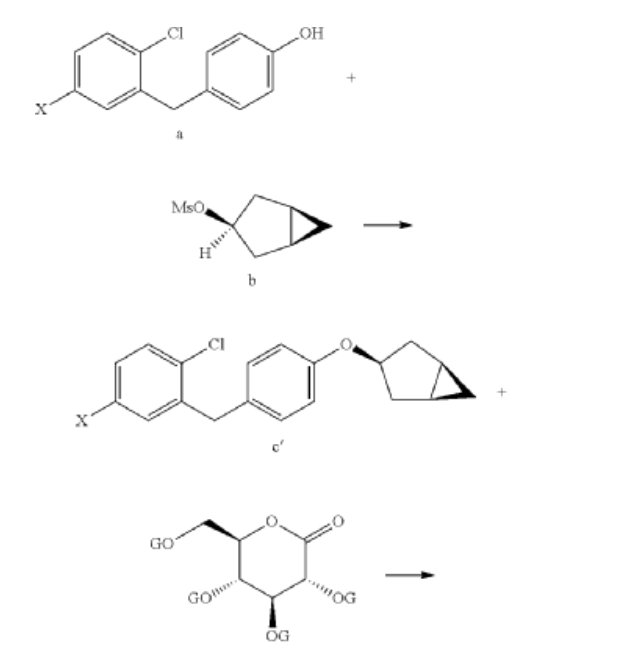
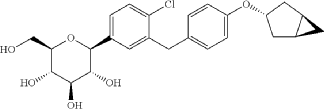
Preparation of (2S,3R,4R,5S,6R)-2-(3-(4-(((1R,3s,5S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Formula II)
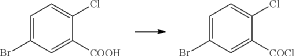
(1) Preparation of 5-bromo-2-chlorobenzoyl chloride
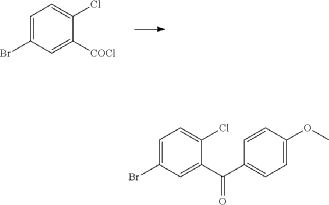
(2) Preparation of (5-bromo-2-chlorophenyl)(4-methoxyphenyl)methanone
(3) Preparation of 4-bromo-1-chloro-2-(4-methoxybenzyl)benzene
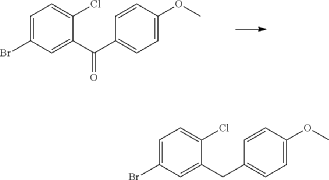
(4) Preparation of 4-(5-bromo-2-chlorobenzyl)phenol
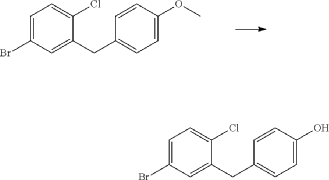
(5) Preparation of (1R,3r,5S)-bicyclo[3.1.0]hexan-3-ol
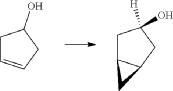
(6) Preparation of (1R,3r,5S)-bicyclo[3.1.0]hexan-3-yl methanesulfonate
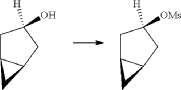
(7) Preparation of (1R,3s,5S)-3-(4-(5-bromo-2-chlorobenzyl)phenyloxy)bicyclo[3.1.0]hexane
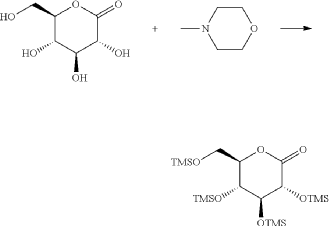
(8) Preparation of (3R,4S,5R,6R)-3,4,5-tri((trimethylsilyl)oxy)-6-(((trimethylsilyl)oxy)methyl)tetrahydro-2H-pyran-2-one
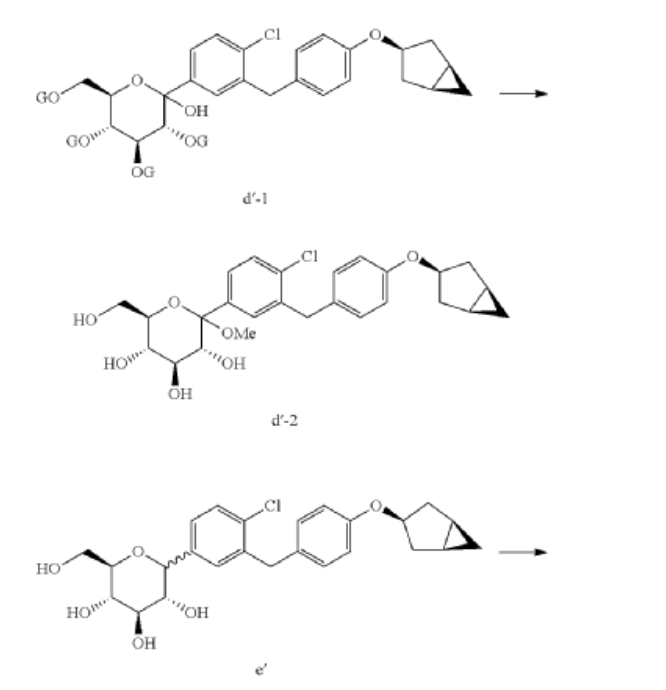
(9) Preparation of (3R,4S,5S,6R)-2-(3-(4-(((1R,3s,5S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol
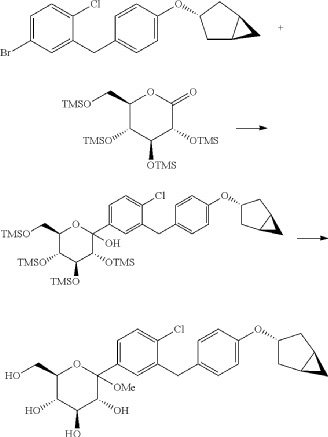
(10) Preparation of (3R,4R,5S,6R)-2-(3-(4-(((1R,3s,5S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
(11) Preparation of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(3-(4-(((1R,3s,5S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
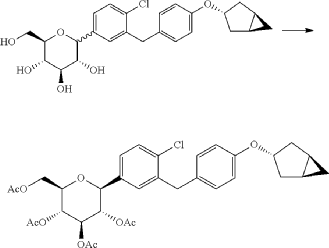
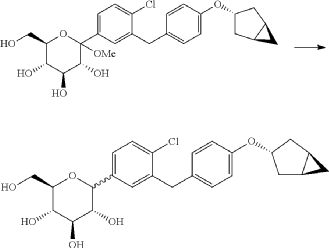
(12) Preparation of (2S,3R,4R,5 S,6R)-2-(3-(4-(((1R,3s,5S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
PAT
EP2891654
https://patentscope.wipo.int/search/en/detail.jsf?docId=EP142501978&_cid=P20-MEQIAN-96633-1
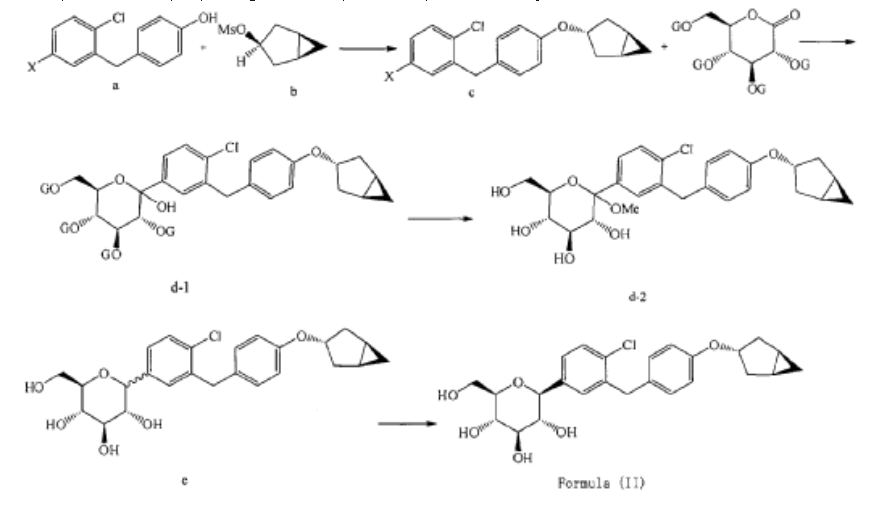
[0027] The compound represented by formula (II) as defined hereinbefore, lab-made, its chemical name and preparation process are described in the following Example 1.
Reference compound 1: Compound 4 as described in the PCT application WO2013/000275A1, lab-made (with reference to the PCT application WO2013/000275A1), its structure is as follows:
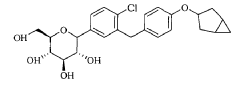
Compound 4, i.e. the compound represented by formula (I).
Reference compound 2: Compound 22 as described in the PCT application WO2013/000275A1, lab-made (with reference to the PCT application WO2013/000275A1), its structure is as follows:
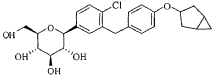
Compound 22.
(12) Preparation of
[0057] (2 S,3 R,4 R,5 S,6 R)-2-(3-(4-(((1 R,3 s,5 S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)-6-(hydr oxymethyl)tetrahydro-2 H-pyran-3,4,5-triol
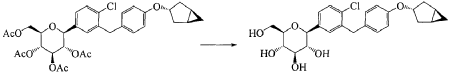
[0058] (2 R,3 R,4 R,5 S,6 S)-2-(acetoxymethyl)-6-(3-(4-(((1 R,3 s,5 S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlo rophenyl)tetrahydro-2 H-pyran-3,4,5-triyl triacetate (81g, 0.129mol) was dissolved in a mixed solvent of tetrahydrofuran (313mL), methanol (470mL) and water (156mL). To the resulting mixture was added lithium hydroxide monohydrate (6.32g, 150mmol). The mixture was stirred at room temperature overnight. TLC indicated the completion of reaction. The solvent was removed from the reaction mixture by rotary evaporation. The residual reaction mixture was dissolved with ethyl acetate (400mL). The organic phase was washed with an aqueous saturated sodium chloride solution, with an aqueous KHSO 4 solution, and with water twice, dried over anhydrous sodium sulphate, and evaporated by rotation. The residue was purified with C18 reverse phase preparative chromatography to produce 54.2g of a final product in a yield of 91%.
Formula: C 25H 29ClO 6 Mw: 460.95 LC-MS( m/ z): 478.3 [M+NH 4] +
1H-NMR (400MHz, MeOD) δ: 7.35-7.26 (m, 3H), 7.08-7.06 (d, 2H), 6.76-6.74 (d, 2H), 4.45-4.41 (m, 1H), 4.10-4.00 (m, 3H), 3.89-3.88 (d, 1H), 3.71-3.69 (m, 1H), 3.45-3.38 (m, 3H), 3.31-3.26 (m, 1H), 2.34-2.29 (m, 2H), 1.87-1.81 (m, 2H), 1.37-1.33 (m, 2H), 0.43-0.42 (m, 1H), 0.11-0.10 (m, 1H).
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Janagliflozin, engineered by Jilin Huisheng Biopharmaceutical Co., Ltd., a subsidiary under the umbrella of Sihuan Pharmaceutical Holdings Group, falls within the category of oral sodium-glucose co-transporter 2(SGLT2) inhibitors. This agent has been specifically designed with the aim of optimizing glycemic regulation in the adult population grappling with type 2 diabetes mellitus (T2DM) [54]. It is marketed under the brand name Huiyoujing. In 2024, the NMPA gave its approval for Janagliflozin, indicated for adult patients with T2DM, where it can be employed either as a standalone treatment (monotherapy) or in combination with metformin to optimize blood glucose regulation [55]. The clinical effectiveness of Janagliflozin was substantiated through a Phase III clinical trial (NCT03811548). This trial specifically assessed its application as a monotherapy in Chinese patients suffering from T2DM
whose blood glucose was not well – managed via diet and exercise alone. The findings of the study indicated notable decreases in glycated hemoglobin levels. Concurrently, improvements were observed in both body weight and blood pressure. Collectively, these outcomes serve as evidence of the drug’s ability to enhance glycemic regulation [56]. Regarding safety, Janagliflozin was generally well-tolerated. In line with the well-established safety characteristics of SGLT2 inhibitors, the frequently encountered adverse events associated with this treatment were urinary tract infections and genital mycotic infections. No serious adverse events were reported during the trial [57].
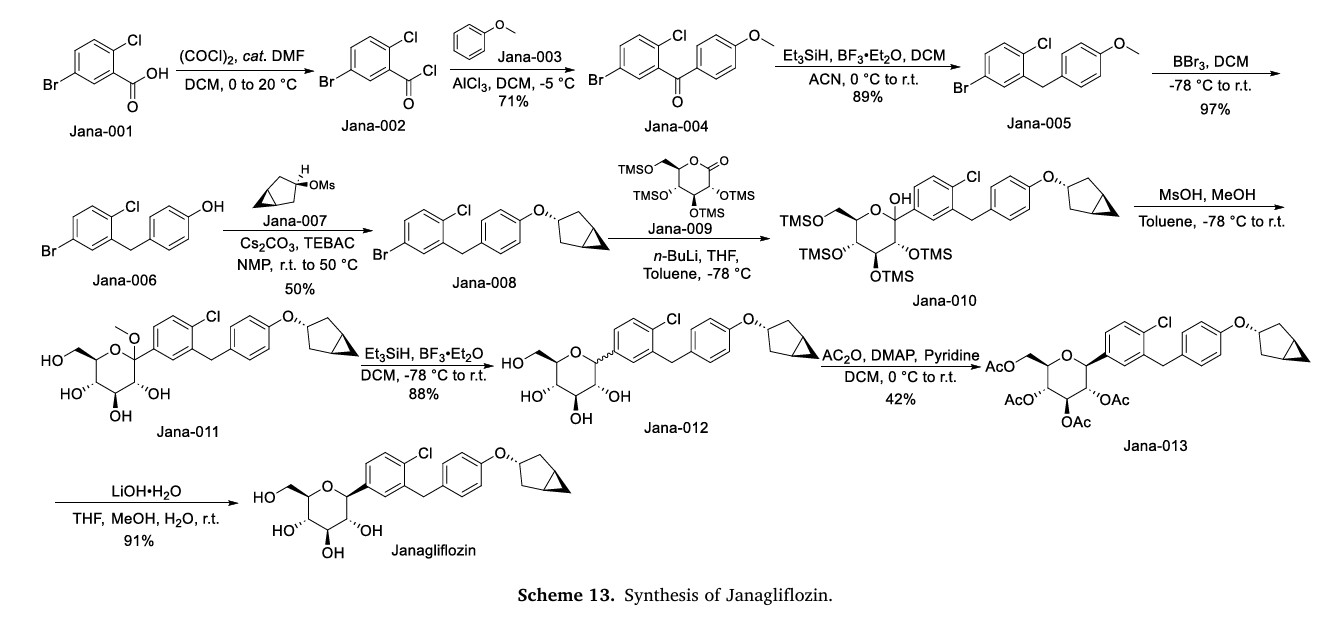
The synthesis of Janagliflozin, depicted in Scheme 13, commences with the acylation of 5-bromo-2-chlorobenzoic acid (Jana-001) using oxalyl chloride, yielding the acyl chloride intermediate Jana-002 [58]. Friedel-Crafts acylation of Jana-002 with anisole (Jana-003) affords ketone Jana-004. Subsequent reduction of the carbonyl group in Jana-004 produces Jana-005. Demethylation of Jana-005 with BBr3
generates phenol Jana-006, which undergoes substitution with intermediate Jana-007 to form ether Jana-008. Addition of gluconolactone (Jana-009) to Jana-008 affords Jana-010, where concurrent TMS
deprotection during etherification yields Jana-011. Reduction of Jana-011 using Et3SiH/BF3.ET2Oproduces Jana-012which is sequentially esterified with Ac2O , and hydrolyzed under LiOH conditions, ultimately yielding Janagliflozin
[54] L. Gao, Z. Cheng, B. Su, X. Su, W. Song, Y. Guo, L. Liao, X. Chen, J. Li, X. Tan, F. Xu,
S. Pang, K. Wang, J. Ye, Y. Wang, L. Chen, J. Sun, L. Ji, Efficacy and safety of
janagliflozin as add-on therapy to metformin in Chinese patients with type 2
diabetes inadequately controlled with metformin alone: a multicentre,
randomized, double-blind, placebo-controlled, phase 3 trial, Diabetes Obes Metab
25 (2023) 785–795.
[55] L. Ji, X. Jiang, Q. Hao, Z. Cheng, K. Wang, S. Pang, M. Liu, Y. Guo, X. Chen, X. Su,
T. Ning, J. Liu, F. Bian, Y. Li, Z. Zhang, W. Song, J. Sun, Efficacy and safety of
janagliflozin monotherapy in Chinese patients with type 2 diabetes mellitus
inadequately controlled on diet and exercise: a multicentre, randomized, double-
blind, placebo-controlled, phase 3 trial, Diabetes Obes Metab 25 (2023)
1229–1240.
[56] L. Song, X. Wang, J. Sun, X. Hu, H. Li, P. Hu, D. Liu, A model-informed approach to
accelerate the clinical development of janagliflozin, an innovative SGLT2 inhibitor,
Clin. Pharmacokinet. 62 (2023) 505–518.
[57] Canagliflozin, Drugs and Lactation Database (Lactmed®), National Institute of
Child Health and Human Development, Bethesda (MD), 2006.
[58] F. Wu, Optically Pure benzyl-4-chlorophenyl-C-glucoside Derivatives as SGLT
Inhibitors (Diabetes Mellitus), 2015. EP2891654.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- Song, Ling; Yao, Xueting; Liu, Yang; Zhong, Wen; Jiang, Ji; Liu, Hongzhong; Zhou, Huimin; Shi, Chongtie; Zong, Kaiqi; Wang, Chong; Ma, Chuanxiang; Liu, Dongyang; Hu, Pei (April 2020). “Translational prediction of first-in-human pharmacokinetics and pharmacodynamics of janagliflozin, a selective SGLT2 inhibitor, using allometric scaling, dedrick and PK/PD modeling methods”. European Journal of Pharmaceutical Sciences. 147: 105281. doi:10.1016/j.ejps.2020.105281. S2CID 212405270.
- Liu, Dongyang; Song, Ling; Wang, Xiaoxu; Liu, Xu; Cao, Fangrui; Liu, Hongzhong; Ding, Yanhua; Xiao, Xinhua; Jiang, Ji; Hu, Pei (1 June 2019). “154-LB: Accelerating Clinical Development of Janagliflozin, a Novel Antidiabetic Drug, Using Model-Informed Drug Development Strategy”. Diabetes. 68 (Supplement_1). doi:10.2337/db19-154-LB. S2CID 195440798.
- Zhao, Hengli; Wei, Yilin; He, Kun; Zhao, Xiaoyu; Mu, Hongli; Wen, Qing (December 2022). “Prediction of janagliflozin pharmacokinetics in type 2 diabetes mellitus patients with liver cirrhosis or renal impairment using a physiologically based pharmacokinetic model”. European Journal of Pharmaceutical Sciences. 179: 106298. doi:10.1016/j.ejps.2022.106298. PMID 36162752. S2CID 252505056.
- Zhao, Hengli; Zhao, Zhirui; He, Kun; Mi, Nianrong; Lou, Kai; Dong, Xiaolin; Zhang, Wenyu; Sun, Jingfang; Hu, Xinyu; Pang, Shuguang; Cheng, Hong; Wen, Qing (August 2023). “Pharmacokinetics, Pharmacodynamics and Safety of Janagliflozin in Chinese Type 2 Diabetes Mellitus Patients with Renal Impairment”. Clinical Pharmacokinetics. 62 (8): 1093–1103. doi:10.1007/s40262-023-01256-0. PMID 37284974. S2CID 259097798.
- Gao, Leili; Cheng, Zhifeng; Su, Benli; Su, Xiuhai; Song, Weihong; Guo, Yushan; Liao, Lin; Chen, Xiaowen; Li, Jiarui; Tan, Xingrong; Xu, Fangjiang; Pang, Shuguang; Wang, Kun; Ye, Jun; Wang, Yuan; Chen, Lili; Sun, Jingfang; Ji, Linong (March 2023). “Efficacy and safety of janagliflozin as add‐on therapy to metformin in Chinese patients with type 2 diabetes inadequately controlled with metformin alone: A multicentre, randomized, double‐blind, placebo‐controlled, phase 3 trial”. Diabetes, Obesity and Metabolism. 25 (3): 785–795. doi:10.1111/dom.14926. PMID 36433709. S2CID 253967474.
- Ji, Linong; Jiang, Xiaozhen; Hao, Qingshun; Cheng, Zhifeng; Wang, Kun; Pang, Shuguang; Liu, Meiying; Guo, Yushan; Chen, Xiaowen; Su, Xiuhai; Ning, Tao; Liu, Jie; Bian, Fang; Li, Yulan; Zhang, Zhinong; Song, Weihong; Sun, Jingfang (May 2023). “Efficacy and safety of janagliflozin monotherapy in Chinese patients with type 2 diabetes mellitus inadequately controlled on diet and exercise: A multicentre, randomized, double‐blind, placebo‐controlled, Phase 3 trial”. Diabetes, Obesity and Metabolism. 25 (5): 1229–1240. doi:10.1111/dom.14971. PMID 36594724. S2CID 255474211.
- “NMPA approves China’s second homegrown SGLT2 inhibitor janagliflozin”. bioworld.com. January 23, 2024.
| Legal status | |
|---|---|
| Legal status | Rx in China; investigational elsewhere |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1800115-22-3 |
| PubChem CID | 91820686 |
| DrugBank | DB16209 |
| UNII | WK4RT85HCA |
| Chemical and physical data | |
| Formula | C25H29ClO6 |
| Molar mass | 460.95 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
{kind=link}
///////////Janagliflozin, china 2024, approvals 2024, Jilin Huisheng Biopharmaceutical Co, sihuan, SGLT2 inhibitors, Huiyoujing, WK4RT85HCA, XZP 5695, UNII-WK4RT85HCA, 1800115-22-3
SYN
SYNTHESIS 2024, 56, 906–943
synthesis of janagliflozin (14) was achieved through an eleven-step process in an overall yield of 3% (Scheme 45).71 Initially, the two advanced intermediates were synthesized and then coupled under cryogenic conditions using nBuLi. The construction of 242 commences with the reaction of 5-bromo-2-chlorobenzoic acid (26c) with oxalyl chloride and a catalytic amount of DMF in DCM, yielding the acid
chloride derivative 26c′. This intermediate is then subjected to Friedel–Crafts acylation with anisole to produce 240 in 71% yield. Subsequent reduction of 240 was carried out using boron trifluoride–diethyl etherate and triethylsilane in a DCM/acetonitrile mixture, leading to the formation of 241 in an excellent yield. Demethylation of compound 241 is accomplished using boron tribromide at low temperature, re
sulting in 242 with a yield of 97%. On the other hand, the synthesis of 245 involves two steps starting from commercially available cyclopent-3-en-1-ol (243). The Simmons Smith cyclopropanation of 243 is performed using a mixture of trifluoroacetic acid, diiodomethane, and diethylzinc in DCM, providing 244 with a yield of 48%. Compound 244 is then further treated with methanesulfonyl chloride to
give the mesylated compound 245 in a yield of 68%. Subsequently, 4-(5-bromo-2-chlorobenzyl)phenol (242) is allowed to react with 245 in the presence of NMP, cesium carbonate, and BTEAC (benzyltriethylammonium chloride) to give 246. The next step involves a lithium–halogen exchange on
246 using n-butyllithium, with addition to 22 at –78 °C affording the hydroxy intermediate. Methylation of this hydroxy intermediate using methanesulfonic acid and methanol provides 247 in 98% yield. Reduction of 247 using boron trifluoride–diethyl etherate and triethylsilane at –78 °C furnishes 248. To achieve the desired isomer, all of the hydroxy groups of compound 248 were protected using acetic anhydride, DMAP, and pyridine in DCM at 0 °C to give the O-acylated compound 249. In the final step, 249 is hydrolyzed us ing lithium hydroxide monohydrate in a mixed solvent consisting of methanol, THF, and water to provide the desired compound janagliflozin (14) in a yield of 91%. This truncated synthetic route is protection-group-free, and is well suited for scale-up. The drawback of the synthetic route is
the late-stage enrichment of the desired isomer in the final product via acylated derivative 249. The poor isolated yield of 249 is not commercially favored due to low throughput and an increase in raw material and production costs

(71) (a) Wu, F. US9315438B2, 2016. (b) Wu, F. EP2891654A1, 2014.