














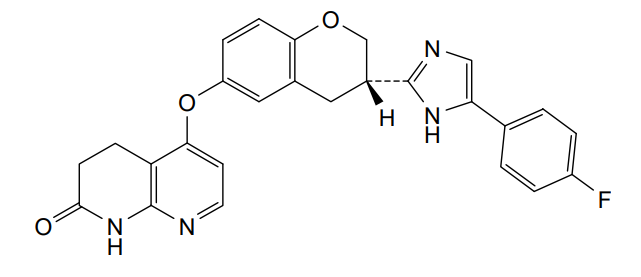
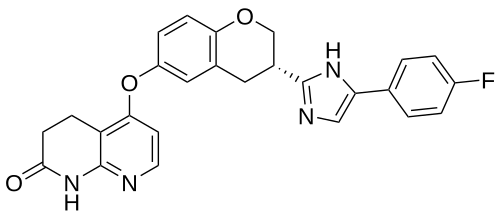
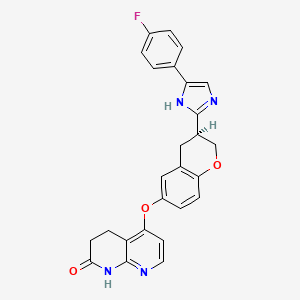
Flezurafenib
CAS 2760321-00-2
MF C26H21FN4O3 MW456.5 g/mol, P26TTM6U27
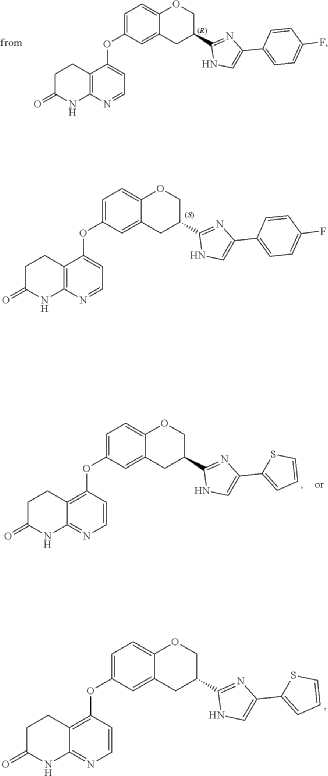
5-({(3S)-3-[4-(4-fluorophenyl)-1H-imidazol-2-yl]-3,4-dihydro-2H-1-benzopyran-6-yl}oxy)-3,4-dihydro-1,8-naphthyridin-2(1H)-one
5-[[(3S)-3-[5-(4-fluorophenyl)-1H-imidazol-2-yl]-3,4-dihydro-2H-chromen-6-yl]oxy]-3,4-dihydro-1H-1,8-naphthyridin-2-one
rapidly accelerated fibrosarcoma (Raf) kinase inhibitor,
antineoplastic
Flezurafenib is an investigational new drug designed as a rapidly accelerated fibrosarcoma (RAF) kinase inhibitor which is being evaluated for the treatment of cancer. Developed by Jazz Pharmaceuticals, this novel therapeutic agent is currently being explored for its efficacy against solid tumors and hematological malignancies harboring oncogenic mutations that activate the RAS-RAF-MAPK signaling pathway.[1][2] As of January 2025, flezurafenib has reached Phase 1 clinical trials, where it is being evaluated for the treatment of advanced cancers and advanced malignant solid neoplasms.[1]
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022023450&_cid=P11-MGN3DV-58095-1
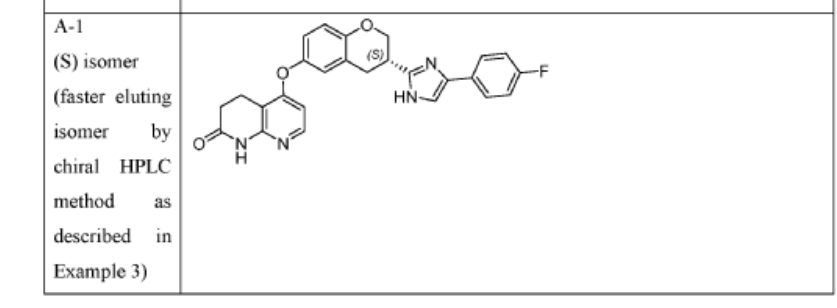
[0402] Example 3. Chiral Synthesis of Compounds A-l and A-2
[0403] A. Synthesis of P2
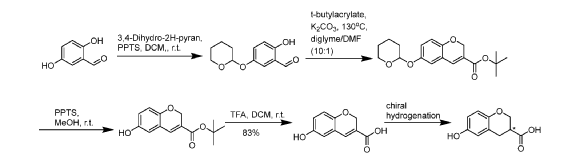
[0404] Step 1: To a solution of 2,5-dihydroxybenzaldehyde (200 g, 1448 mmol) and pyridinium p-toluenesulfonate (18.2 g, 72.4 mmol) in DCM (3.75 L) was added 3,4-dihydro-2H-pyran (165 mL, 1810 mmol) dropwise over 10 minutes and the reaction temperature warmed to 30 °C. The reaction was stirred for 2 hours and checked by UPLC-MS which indicated the reaction was 92% complete (~5% starting material and ~3% later running unknown). The reaction was stopped. The reaction was washed with water (1.5 L) and the DCM solution was passed through a 750g silica pad and followed through by DCM (2.5 L). The DCM solution was reduced in-vacuo and the crude product was then slowly diluted with Pet. Ether to ~1L total volume, stirred and cooled to -10° C to afford a thick yellow slurry. The product was filtered and washed with Pet. Ether (2 x 150 mL) and pulled dry for 3 hours to afford 2-hydroxy-5-tetrahydropyran-2-yloxy-benzaldehyde (265g, 1192 mmol, 82% yield) as a bright yellow solid. ¾ NMR (400 MHz, DMSO-d6) d/ppm: 10.35 (s, 1H), 10.23 (s, 1H), 7.32 – 7.19 (m, 2H), 6.94 (d, J = 8.9 Hz, 1H), 5.36 (t, J = 3.3 Hz, 1H), 3.77 (ddd, J = 11.2, 8.8, 3.6 Hz, 1H), 3.59 – 3.49 (m, 1H), 1.94 – 1.45 (m, 6H). UPLC-MS (ES+, Short acidic): 1.64 min, m/z 223.0 [M+H]+ (100%).
[0405] Step 2: 2-hydroxy-5-tetrahydropyran-2-yloxy-benzaldehyde (107 g, 481 mmol) was dissolved in diglyme (750 mL) and K2CO3 (133 g, 963 mmol) was added on one portion with stirring to afford a bright yellow suspension. The reaction was then heated to 140°C and tert-butyl acrylate (155 mL, 1059 mmol) in DMF (75 mL) was added over 10 minutes starting at ~110°C and up to 130°C. Maintained this temperature for a further 1 hour. UPLC-MS indicated that the
reaction had progressed 75%. After a further hour this showed clean conversion to 85% product and little or no side-products. After another 3 hours UPLC-MS showed 88% product (previous reactions had showed that further heating did not afford more conversion). The dark brown reaction was cooled to room temperature overnight and filtered to remove inorganics. The reaction was suspended in EtOAc (2.5 L) and water (2.5 L) and the phases separated. The aqueous was re-extracted with EtOAc (2.5 L) and the combined organics were washed with brine (2 x 1.5 L) and the organics were reduced in-vacuo. The crude product was then purified on silica (2Kg) loading in a minimum volume of DCM. A gradient of EtOAc in Pet. Ether (10 – 25%) was run and clean product fractions combined and reduced in-vacuo to afford tert-butyl 6-tetrahydropyran-2-yloxy-2H-chromene-3-carboxylate (93.5 g, 281 mmol, 58% yield) as a yellow solid. ¾ NMR (400 MHz, DMSO-d6) d/ppm: 7.37 (q, J = 1.2 Hz, 1H), 7.05 (d, J = 2.9 Hz, 1H), 6.94 (dd, J = 8.8, 2.9 Hz, 1H), 6.79 (dd, J = 8.7, 0.7 Hz, 1H), 5.35 (t, J = 3.3 Hz, 1H), 4.82 (d, J = 1.4 Hz, 2H), 3.77 (ddt, J = 13.3, 8.3, 4.2 Hz, 1H), 3.59 – 3.48 (m, 1H), 1.93 – 1.49 (m, 6H), 1.49 (s, 9H). UPLC-MS (ES+, Short acidic): 2.18 min, m/z ([M+H]+) not detected (100%).
[0406] Step 3: tert-butyl 6-tetrahydropyran-2-yloxy-2H-chromene-3-carboxylate (215 g, 647 mmol) was suspended in MeOH (1.6 L) at room temperature (did not dissolve immediately) and pyridinium p-toluenesulfonate (16.3 g, 64.7 mmol) added. The reaction was warmed to 40°C with a hot water bath and checked by UPLC-MS for progress after 1 hour which indicated the reaction was complete and was a clear orange solution. The reaction was reduced in-vacuo and the crude product dissolved in DCM (2 L) and washed with water (1 L). The organic layer was dried (MgSC>4), filtered and reduced in-vacuo to afford the crude product as a yellow solid. This was suspended in Pet. Ether and stirred in an ice bath before filtering, to afford a bright yellow solid. This was dried under high vac at 50°C for 2 hours to afford tert-butyl 6-hydroxy-2H-chromene-3-carboxylate (144.4 g, 582 mmol, 90% yield). ¾ NMR (400 MHz, DMSO-d6) d/ppm: 9.17 (s, 1H), 7.33 (s, 1H), 6.76 – 6.64 (m, 3H), 4.77 (d, J = 1.4 Hz, 2H), 1.49 (s, 9H). UPLC-MS (ES+, Short acidic): 1.71 min, m/z 247.2 [M-H]- (100%).
[0407] Step 4: tert-Butyl 6-hydroxy-2H-chromene-3-carboxylate (84. g, 338.34mmol) was dissolved in DCM (500mL) and trifluoroacetic acid (177.72mL, 2320.9mmol) added at room temperature and the reaction stirred to give a brown solution. Initially gas evolution was noted and the reaction was stirred over several days at room temperature. DCM and TFA were removed in-vacuo and finally azeotroped with 200ml of toluene before slurrying with diethyl ether and filtering to give the crude product 6-hydroxy-2H-chromene-3-carboxylic acid (53.15g, 276.58mmol, 81.745% yield) as a cream solid. ¾ NMR (400 MHz, DMSO-d6) d/ppm: 12.77 (s, 1H), 9.14 (s, 1H), 7.37 (t, J = 1.4 Hz, 1H), 6.72 (dd, J = 2.4, 0.9 Hz, 1H), 6.70 – 6.64 (m, 2H), 4.78 (d, J = 1.4 Hz, 2H).
[0408] Step 5: (R)-Phanephos and [RuCl2(p-cym)]2 (1.2: 1 eq., 6.6 mg, 3.0 mg respectively) were weighed into a 50 mL glass lined Parr vessel followed by the substrate (1.845 g, 9.6 mmol). Methanol (16 mL, 0.6 M substrate concentration) was added to the vessel followed by triethylamine (135 μL, 0.96 mmol, 0.1 eq.). A PTFE stirrer bar was added and the thermocouple was covered with PTFE tape. The vessel was sealed and purged with nitrogen 5 times (at ~2 bar) and 5 times with stirring (~500 rpm). The vessel was then purged with hydrogen 5 times (at -10 bar) and 5 times with stirring (~500 rpm). The vessel was then pressurised to 5 bar hydrogen pressure and heated to 40 °C (with 1500 rpm stirring speed). The pressure was kept constant but with venting and refilling to 5 bar after sampling. After 21.5 hours, the vessel was allowed to cool. After 22.5 hours, the vessel was vented and purged with nitrogen. Each -0.1 mL sample was diluted to -1 mL with MeOH for SFC analysis. Work-up procedure: MeOH removed by concentrating under vacuum, followed by addition of EtOAc (10 mL) and 1 M HC1 (10 mL). The layers were mixed before separating. The EtOAc layer was washed with a further portion of 1 M HC1 (4 mL) before removing the aqueous layer to leave the EtOAc organic phase. The aqueous layer was then washed with a further portion of EtOAc (4 mL) and the organic layers were combined. EtOAc was then removed under vacuum to leave behind the product as a greyish solid (See Table 29). P2 is the first eluting product with a retention time of 5.8 min and PI is the second eluting product with a retention time of 6.1 min using the SFC method as described in Example 1.
[0409] B. Synthesis of 5-fluoro-3,4-dihydro-l,8-naphthyridin-2(lH)-one
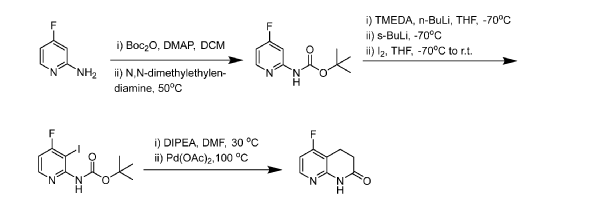
0410] Step 1: 2-Amino-4-fluoropyridine (400 g, 3568 mmol) was charged into a 10 L fixed reactor vessel and then taken up in DCM (4 L) as a slurry under nitrogen atmosphere. To this was added DMAP (43.6 g, 357 mmol) and cooled to 10°C. Di-tert-butyldicarbonate (934 g, 4282 mmol) was added, as a solution in DCM (1 L), over the space of 1.5 hours. The reaction was stirred at room temperature for 2 hours after which time the complete consumption of the starting material was evident by NMR. To the reaction was added N,N-dimethylethylenediamine (390 mL, 3568 mmol) and the reaction warmed to 40°C overnight (converting any di-BOC material back to the mono-BOC desired product). Allowed to cool to room temperature and then diluted with further DCM (2 L) and washed with water (2 L). Extracted with further DCM (2 L), washed with water
(1 L), brine (1.2 L) and dried (MgSO4) before filtering. The solvents were removed in-vacuo and the resultant product was slurried in DCM/Pet. Ether (1:1) (500 mL). Filtered, washed with further Pet. Ether and pulled dry to afford tert- butyl N-(4-fluoro-2-pyridyl)carbamate (505 g, 2380 mmol, 67% yield) as a cream solid product. A second crop of material was isolated from the mother liquors after passing through a short pad of silica followed by trituration with DCM/Pet. Ether (1:1) (-200 mL) to afford tert-butyl N-(4-fluoro-2-pyridyl)carbamate (46.7 g, 220 mmol, 6% yield). ¾ NMR (400 MHz, DMSO-d6) d/ppm: 10.13 (d, J = 1.7 Hz, 1H), 8.26 (dd, J = 9.4, 5.7 Hz, 1H), 7.60 (dd, J = 12.3, 2.4 Hz, 1H), 6.94 (ddd, J = 8.2, 5.7, 2.4 Hz, 1H), 1.47 (s, 9H). UPLC-MS (ES+, Short acidic): 1.64 min, m/z 213.1 [M+H]+ (98%).
[0411] Step 2: tert-butyl-N-(4-fluoro-2-pyridyl)carbamate (126 g, 594 mmol) and TMEDA (223 mL, 1484 mmol) were taken up in dry THF (1.7 L) and then cooled to -78°C under nitrogen atmosphere. To this solution was added n-butyllithium solution (2.5M solution in hexanes) (285 mL, 713 mmol) and then allowed to stir for a further 10 minutes. sec-Butyllithium solution (1.2M in cyclohexane) (509 mL, 713 mmol) was added keeping the reaction temperature below -70°C whilst stirred for 1 hour. After this time, Iodine (226 g, 891 mmol) in THF (300 mL) was added slowly and dropwise over 30 minutes to keep the temp below -65°C. Stirred at -70°C for another 10 minutes and then quenched by the addition of sat. aq. NH4CI solution (400 mL) and then a solution of sodium thiosulphate (134 g, 848 mmol) dissolved in water (600 mL). This addition raised the temperature to — 25°C. The reaction was warmed to room temperature then transferred to the 5L separator and extracted with EtOAc (2 x 1.5 L) and then washed with brine (500 mL), dried (MgSCL) and then evaporated in vacuo to afford crude material (~200g). This was taken up in hot DCM (500 mL) (slurry added to the silica pad) and then passed through a 2Kg silica pad. Washed through with DCM (10 x 1 L fractions) and then the product was eluted from the column with EtOAc in Pet. Ether (10% to 100%), (1 L at each 10% increase, with 1 L fractions). This gave 2 mixed fractions and clean product containing fractions, which were combined and evaporated in vacuo to afford tert-butyl N-(4-fluoro-3-iodo-2-pyridyl)carbamate (113.4 g, 335.4 mmol, 57% yield) as a white solid. Clean by UPLC-MS and NMR. The mixed fractions were combined with previous crude material to afford 190g in total of a cream solid that was composed of -50% of the desired product. This was re-columned as above to afford a combined second crop from all 4 batches as a cream solid tert-butyl N-(4-fluoro-3-iodo-2-pyridyl) carbamate (107.5 g, 318 mmol, 54% yield). ¾ NMR (400 MHz, DMSO-d6) d/ppm: 9.47 (s, 1H), 8.33 (dd, J = 8.7, 5.5 Hz, 1H), 7.19 (dd, J = 7.3, 5.5 Hz, 1H), 1.46 (s, 9H). UPLC-MS (ES+, Short acidic): 1.60 mm, m/z 339.1 [M+H]+ (100%).
[0412] Step 3: tert-butyl N-(4-fluoro-3-iodo-2-pyridyl)carbamate (300 g, 887 mmol), 3,3-dimethoxyprop- 1 -ene (137 mL, 1153 mmol) and DIPEA (325 mL, 1863 mmol) were suspended in DMF (2 L) and water (440 mL) to give a yellow slurry. This was degassed for 20 minutes at 30°C. To this mixture was then added Palladium (II) acetate (19.92 g, 89 mmol) in one portion and degassed again for a further 15mins. The reaction was slowly and carefully heated to 100°C. Gas evolution at around 85°C (large volumes of off gassing, presumably due to the loss of Boc group as CO2 and isobutylene). The reaction became darker once off gassing finished and full solubility achieved. The reaction was then heated at 100°C for 3 hours and checked by UPLC-MS (70% desired product, 18% un-cyclised intermediate and 7% des-iodo BOC). The reaction was heated for a further 2 hours and this showed 81% desired product, 12% un-cyclised intermediate and 8% des-iodo BOC. After 7 hours the reaction showed 89% desired product, 4% un-cyclised
intermediate and 7% des-iodo BOC. The reaction was heated overnight. The reaction solution was cooled and filtered through celite and evaporated in-vacuo to a thick dark orange slurry which was then suspended in water (1 L) and acidified to pH~l-2 with aq. HC1 (4N) solution. This was then basified to pH~9 with sat. aq. Na2CO3 solution. Extracted with DCM (2 x 2L) and washed with brine and dried (MgS04). EtOAc (2 L) was added to the solution and then the organics were passed through a 500g silica plug. This was then followed by DCM/EtOAc (1 : 1) (2 L) and finally EtOAc (2 L) (the final wash through contained only baseline). The product containing fractions were combined and reduced in-vacuo to give an orange slurry and then suspended in hot diethyl ether (300 mL), cooled back to ~10°C in an ice bath with stirring before being filtered and washed with 150 mL of ice cold diethyl ether. Pulled dry to afford 5-fluoro-3,4-dihydro-lH-l,8-naphthyridin- 2-one (58.4 g, 351.5 mmol, 39.6 % yield) as a cream fluffy solid. ¾ NMR (400 MHz, DMSO-d6) d/ppm: 10.69 (s, 1H), 8.29 – 7.90 (m, 1H), 6.92 (dd, J = 8.8, 5.7 Hz, 1H), 2.88 (dd, J= 8.3, 7.1 Hz, 2H), 2.57 – 2.47 (m, 2H). UPLC-MS (ES+, Short acidic): 1.04 mm, m/z 167.0 [M+H]+ (100%).
[0413] C. Synthesis of Compounds A-l and A-2
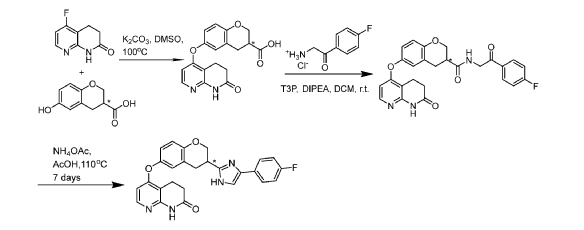
[0414] Step 1: Potassium carbonate (832mg, 6.02mmol) was added to a stirred solution of 5- fluoro-3,4-dihydro-lH-l,8-naphthyridin-2-one (250mg, 1.5mmol), P2 (see step A, 292mg, 1.5mmol; 85% ee) and DMSO (2mL) at room temperature. The reaction was degassed and flushed with nitrogen 3 times before being stirred under a nitrogen atmosphere for 18 hours at 100°C. The reaction mixture was cooled to room temperature and diluted with water (20mL) and the resulting mixture extracted with EtOAc (20mL). A solution of citric acid (1156.3mg, 6.02mmol) in water (lOmL) was then added to the aqueous layer resulting in a solid precipitate which was filtered and dried in vacuo to give (S)- or (R)-6-[(7-oxo-6, 8-dihydro- 5H-1 ,8-naphthyridin-4-yl)oxy]chromane-3 -carboxylic acid (345mg, 1.01 mmol, 67% yield) as a white solid. UPLC-MS (ES+, Short acidic): 1.29 mm, m/z 341.1 [M+H]+. ¾ NMR (400 MHz, DMSO-d6) d/ppm: 12.71 (lH, br s), 10.47 (1H, s), 7.95 (1H, d, J = 6.0Hz), 6.97 (1H, d, J = 2.4Hz), 6.89 (1H, dd, J = 8.4Hz, 2.4Hz), 6.83 (1H, d, J = 8.4Hz), 6.24 (1H, d, J = 6.0Hz), 4.33 (1H, dd, J = 11.2Hz, 3.2Hz), 4.15 (1H, dd, J = 11.2Hz, 7.2Hz), 3.05-2.89 (5H, m), 2.53 (2H, t, J = 7.6Hz).
[0415] Step 2: Propylphosphonic anhydride (0.91mL, 1.52mmol) was added to a stirred solution of (S)-6-[(7-oxo-6,8-dihydro-5H-l,8-naphthyridin-4-yl)oxy]chromane-3-carboxylic acid (345mg, 1.01 mmol), 2-amino- l-(4-fluorophenyl)ethanone hydrochloride (288mg, 1.52mmol), N,N-diisopropylethylamine (0.88mL, 5.07mmol) andDCM (lOmL) at room temperature. After stirring for 2 hours the reaction was complete by LCMS. Water (50mL) and DCM (50mL) were added and the organic layer separated and washed with sat. aq. Na2CO3 (50mL). The organic layer was dried over sodium sulfate and solvent removed in vacuo. The residue was purified by column chromatography using an eluent of 0-5% MeOH in DCM to give (S)- or (R)-N-[2-(4-fluorophenyl)-2-oxo-ethyl]-6-[(7-oxo-6,8-dihydro-5H-l,8-naphthyridin-4-yl)oxy]chromane-3-carboxamide (300mg, 0.63mmol, 62% yield) as a yellow solid. UPLC-MS (ES+, Short acidic): 1.52 mm, m/z 476.4 [M+H]+. ¾ NMR (400 MHz, DMSO-d6) d/ppm: 10.47 (1H, s), 8.60-8.54 (1H, m), 8.08 (1H, dd, J = 8.8Hz, 5.6Hz), 7.95 (1H, d, J = 5.6Hz), 7.41-7.37 (2H, m), 7.01-6.97 (1H, m), 6.90 (1H, dd, J = 8.8Hz, 3.2Hz), 6.86 (1H, d, J = 8.8Hz), 6.25 (1H, d, J = 5.6Hz), 4.65 (2H, d, J = 6.0Hz), 4.42-4.35 (1H, m), 3.96 (1H, t, J = 9.6Hz), 3.03-2.87 (5H, m), 2.55-2.52 (2H, m), 1 exchangeable proton not seen.
[0416] Step 3: (S)- or (R)-N-[2-(4-fluorophenyl)-2-oxo-ethyl]-6-[(7-oxo-6, 8-dihydro- 5H-1, 8-naphthyridin-4-yl)oxy]chromane-3 -carboxamide (300mg, 0.63mmol), ammonium acetate
(1216mg, 15.77mmol) and acetic acid (5mL) were combined in a sealable vial, the vial sealed and the reaction stirred and heated to 130°C for 18 hours after which time the reaction was complete by LCMS. The reaction was cooled to room temperature and AcOH removed in vacuo. DCM (50mL) was added to the residue and sat. aq. Na2CO3 (50mL) added. The organic layer was separated and washed with brine, dried over sodium sulfate and solvent removed in vacuo. The residue was purified by column chromatography using an eluent of 0-10% MeOH in DCM to give (R)- or (S)-5 – [3 – [4-(4-fluorophenyl)- 1 H-imidazol-2-y 1] chroman-6-yl] oxy-3 ,4-dihydro- 1 H- 1 , 8-naphthyridin-2-one (141mg, 0.31mmol, 49% yield) as a yellow solid.
[0417] Chiral LCMS of the product, together with chiral LCMS’s of Compounds A-l and A-2 showed that this product is predominantly Compounds A-l (Fig. 7), with a similar ee to that of the starting acid (85% ee), however accurate analysis cannot be done due to overlap of the peaks. UPLC-MS (ES+, Short acidic): 1.36 mm, m/z 457.2 [M+H]+. Ή NMR (400 MHz, DMSO-d6) d/ppm: 12.31 (0.2H, s), 12.10 (0.8H, s), 10.47 (1H, s), 7.96 (1H, d, J = 6.0Hz), 7.80-7.75 (1.8H, m), 7.69-7.65 (0.2H, m), 7.59-7.78 (0.8H, m), 7.29-7.23 (0.4H, m), 7.19-7.13 (1.8H, m), 7.03-7.00 (1H, m), 6.92 (1H, dd, J = 8.8Hz, 2.8Hz), 6.89 (1H, d, J = 8.8Hz), 6.27 (1H, d, J = 6.0Hz), 4.55-4.48 (1H, m), 4.16-4.09 (1H, m), 3.44-3.36 (1H, m), 3.30-3.21 (1H, m), 3.16-3.09 (1H, m), 2.94 (2H, t, J = 7.2Hz), 2.54 (2H, t, J = 7.2Hz).
[0439] A. Synthesis of P2
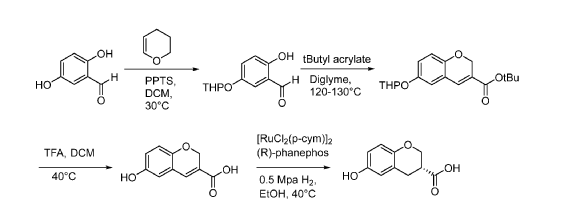
[0440] Step 1: 2,5-Dihydroxybenzaldehyde (13.6 kg, 98.18 mol) was dried using 2 x azeotropic concentrations with 2 x 125-130 kg of THF at up to 35 °C, concentrating under vacuum to 27-41 kg each time. The THF was then removed using 4 x azeotropic concentrations with 4 x 179-187 kg of DCM at up to 35 °C, concentrating under vacuum to 27-41 kg each time. The concentrate was diluted with DCM (284 kg) and pyridine p-toluenesulfonate (PPTS; 1.25 kg, 4.97 mol) was added. 3,4-dihydro-2H-pyran (10.4 kg, 123.63 mol) was added slowly at between 25-35 °C and the reaction was stirred at 30 °C for 90 minutes. The mixture was added to a solution of Na2CO3 (7.1 kg) in water (138 kg) at -15 °C and allowed to warm to 25 °C and then stirred for 6 h. The mixture was filtered through Celite® (33 kg), washing with DCM (92.5 kg). The filtrate was allowed to stand for 1 h and then the organic phase was separated and concentrated to 27-41 kg.
The DCM was then removed using 3 x azeotropic concentrations with 3 x 105 kg n-heptane at up to 35 °C, concentrating under vacuum to 27-41 kg each time. The concentrate was diluted with n- heptane (210 kg) and the heated to 30-40 °C and stirred for 6 h. The solution was then cooled to – 5 to -15 °C over 4 h, stirred for 9 h and filtered, washing the filter cake with n-heptane (39.5 kg).
The wet cake was dried at 30-40 °C for 24 h in vacuo to give 2-hydroxy-5-(oxan-2- yloxy)benzaldehyde (9.38 kg, 40.6%). Additional product (8.00 kg, 34.3%) was recovered by dissolving solid attached to the walls of the reaction vessel with 42 kg DCM and concentrating the resultant solution in vacuo to give a further 8.00 kg (34.3% yield ) of product to give a total yield of 74.9% (17.38 kg). LCMS (ES-): 15.18 mm, m/z 221.12 [M-H]-.
[0441] Step 2: To a stirring solution of 2-hydroxy-5-(oxan-2-yloxy)benzaldehyde (16.95 kg, 76.27 mol) in diglyme (113.4 kg) was added K2CO3 (21.4 kg, 154.83 mol) and the mixture was heated to between 80-90 °C. Tert-butyl prop-2-enoate (20.0 kg, 156.04 mol) was added, and the mixture was heated to between 120-130 °C and stirred for 18 hr. The mixture was cooled and
filtered, and the filter cake washed with EtOAc (80.0 kg). The filtrate was diluted with EtOAc (238.0 kg) and water (338.0 kg) and stirred for 1 hr at 20-30 °C, then stood for 2 hr. The mixture was filtered through Celite® (40.0 kg), and the filter cake washed with EtOAc (84.0 kg). The filtrate was left to stand for 2 hr and the aqueous layer was extracted with EtOAc (312.0 kg), stirring for 1 hr at 0-30 °C and standing for 2 hr. The organic layers were combined and washed with 2 x 345 kg water, stirring at between 20-30 °C for 1 hr and standing for 2 hr for each wash. The combined organics were then concentrated to 182.4 kg maintaining the temperature below 50 °C under vacuum. This gave the product tert-butyl 6-(oxan-2-yloxy)-2H-chromene-3-carboxylate as a 9.3% solution in diglyme/EtOAc (66.9% yield) and was used in the next stage without further isolation. LCMS (ES-): 20.26 mm, m/z 247.12 [M-THP]-.
[0442] Step 3: Tert-butyl 6-(oxan-2-yloxy)-2H-chromene-3-carboxylate (16.9 kg, 50.84 mol) as a 181.8 kg solution in diglyme/EtOAc was concentrated to 68 kg under vacuum at 50 °C. TFA (110.3 kg, 1002.46 mol) was added and the reaction was warmed to 40 °C under nitrogen flow and then stirred for 8 hrs. The mixture was then diluted with DCM (222.0 kg) and cooled to between -5 and -15 °C, and then stirred for 7 hrs. The solid was filtered and the filter cake washed with DCM (67.0 kg). The wet cake was dried for 24 hr under vacuum at between 30-40 °C to give 6-hydroxy-2H-chromene-3-carboxylic acid (8.75 kg, 78.5% yield). LCMS (ES-): 0.85 min, m/z 191.11 [M-H]-.
[0443] Step 4: To a stirring solution of 6-hydroxy-2H-chromene-3-carboxylic acid (7.19 kg, 37.4 mol) in N2-degassed EtOH (60 kg) was added (R)-Phanephos (131 g, 0.227 mol), [RuCl2(p-cym)]2 (70 g, 0.114 mol), and Et3N (5.6 kg, 55.3 mol). The reaction atmosphere was replaced with 3 x N2 and then 3 x H2, adjusting the H2 pressure to between 0.5-0.6 MPa, and then stirred for 18 hrs at 40 °C. The atmosphere was then replaced with 3 x N2 and then 3 x H2, adjusting the H2 pressure to between 0.5-0.6 MPa again and the mixture was stirred for a further 18 hrs.
[0444] The mixture was concentrated in vacuo to ca. 30 kg at no more than 40 °C. The reaction was diluted with MTBE (53 kg) and cooled to between 15-25 °C. 5% Na2CO3 (80 kg) was added dropwise, and the mixture was stirred for 2 hrs and stood for 2 hrs at between 15-25 °C. The aqueous layer was collected and 5% Na2CO3 (48 kg) was added to the organic layer, then stirred for 2 hrs at 15-25 °C and filtered through Celite® (10.0 kg). The wet cake was washed with water (20 kg) and the combined aqueous filtrate and aqueous layer were diluted with IP Ac (129.0 kg). The pH of the mixture was adjusted to 1-3 with dropwise addition of 6 N HC1 (29 kg) at 15-25 °C and stirred for 2 hrs. The mixture was filtered through Celite® (10 kg), washing the filter cake with IP Ac (34 kg) and the filtrate was left to stand for 2 hrs at 15-25 °C. The aqueous layer was then extracted with IP Ac (34 kg) and the combined organic layers were concentrated to ca. 35 kg under vacuum at no more than 40 °C. Me-cyclohexane (21 kg) was added dropwise at 15-25 °C and concentrated to ca. 35 kg under vacuum at no more than 40 °C. Further Me-cyclohexane (20 kg) was added dropwise at 15-25 °C and stirred for 3 hrs. The mixture was then stirred at 40-50 °C for 4 hrs and cooled to 15-25 °C over 3 hrs and then stirred for a further 2 hrs.
[0445] The mixture was then filtered, washing the filter cake with 16.4 kg of IPAc/Me-cyclohexane (1/4, v/v). The wet cake was dried for 24 hrs at 35-45 °C under vacuum to give (3R)-6-hydroxy-3,4-dihydro-2H-l-benzopyran-3-carboxylic acid (5.2 kg, 68.6% yield, chiral purity 95.5%). Further product was isolated by rinsing solid from the reaction vessel wall with EtOH (42 kg) and concentrating to dryness. The resulting solid was suspended in IP Ac (875mL) and Me-cyclohexane (2625mL) and stirred for 5 h at 40 °C and then cooled to 20 °C over 2 h and stirred for 16 h and filtered. The filter cake was then split into 2 equal batches and each batch suspended in IP Ac (912mL) and Me-cyclohexane (2737mL). The resulting mixtures were stirred at 45 °C for 18 h and then filtered and the filter cake dried at 45 °C to give (3R)-6-hydroxy-3,4-dihydro-2H-l-benzopyran-3 -carboxylic acid (1.27 kg, 17% yield, chiral purity 96.2%). LCMS (ES-): 1.74 min, m/z 193.03 [M-H]-.
[0446] Chiral resolution to improve chiral purity:
[0447] (3R)-6-Hydroxy-3,4-dihydro-2H-l-benzopyran-3-carboxylicacid (P2; 5.94 kg, 30.59 mol) (chiral purity =95.5%) was dissolved in IP Ac (138.2 kg) and stirred for 2 hrs at 20-30 °C. The solution obtained was filtered through Celite® (12 kg), washing through with IP Ac (25 kg). In a separate vessel, (S)-(+)-2-phenylglycinol (4.4 kg, 32.07 mol) was dissolved in IP Ac (56 kg), stirring for 1 hr at 40-50 °C. The filtrate was added to this solution over 4 hrs at 40-50 °C, and stirred for 1 hr. The mixture was then stirred for 1 hr at 15-25 °C, and concentrated to ca. 120 kg under vacuum at no more than 40 °C. The concentrate was stirred for 3 hrs at 15-25 °C and filtered, washing through with IP Ac (12 kg) (chiral purity = 96.2%).
[0448] The wet cake was redissolved in EtOH (29 kg), heated to 40-50 °C and diluted with IP Ac (64 kg). 30 g of dry product was added and stirred for 30 min at 15-25 °C. The mixture was concentrated to ca. 42 kg under vacuum at no more than 40 °C, and rediluted with IP Ac (64 kg). This step was repeated two additional times, then stirred at 40-50 °C for 8 hrs. The mixture was filtered, washing through with IP Ac (13 kg) (chiral purity = 97.7%). This recrystallisation process was repeated two further times, for a total of 3 recrystallisation rounds to give material with 98.9% chiral purity.
[0449] The wet cake (10.7 kg) was then dissolved in IN HC1 (45.4 kg) and stirred for 1 hr at 20-30 °C. The mixture was filtered through Celite® (11.5 kg), washing through with IP Ac (28 kg). The aqueous layer was extracted with IP Ac (28.8 kg) and the combined organic layers were washed with water (30 kg), then concentrated to ca. 24 kg at 40 °C under vacuum. Me-cyclohexane (19 kg) was added at 20 °C and the mixture was concentrated to ca. 24 kg at 40 °C under vacuum. This step was repeated twice more. The concentrate was diluted with Me-cyclohexane (29 kg) and stirred for 1 hr at 15-25 °C. The mixture was filtered, and the wet cake was rinsed with Me-Cyclohexane (59 kg). The wet cake was dried under vacuum at 35-45 °C for 16 hrs to give (3R)-6-hydroxy-3,4-dihydro-2H-l-benzopyran-3-carboxylic acid (3.02 kg, 50.2% yield).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US350349340&_cid=P11-MGN37Z-55206-1
PAT
- Methods of treating solid tumors with mitogen activated protein kinase (mapk) pathway alterationsPublication Number: WO-2024173931-A2Priority Date: 2023-02-17
- Crystalline forms of (s)-5-((3-(4-(4-fluorophenyl)-1h-imidazol-2-yl)chroman-6-yl)oxy)-3,4-dihydro-1,8-naphthyridin-2(1h)-onePublication Number: TW-202421110-APriority Date: 2022-11-29
- Crystalline forms of (s)-5-((3-(4-(4-fluorophenyl)-1h-imidazol-2-yl)chroman-6-yl)oxy)-3,4-dihydro-1,8-naphthyridin-2(1h)-onePublication Number: WO-2024115583-A1Priority Date: 2022-11-29
- Chiral synthesis of fused bicyclic raf inhibitorsPublication Number: US-2022041595-A1Priority Date: 2020-07-28
- Chiral synthesis of fused bicyclic raf inhibitorsPublication Number: WO-2022023450-A1Priority Date: 2020-07-28
- Chiral synthesis of fused bicyclic raf inhibitorsPublication Number: EP-4188923-A1Priority Date: 2020-07-28
- Chiral synthesis of fused bicyclic RAF inhibitorsPublication Number: KR-20230058630-APriority Date: 2020-07-28
- Chiral Synthesis of Fused Bicyclic RAF InhibitorsPublication Number: CN-116348465-APriority Date: 2020-07-28
- Chiral Synthesis of Fused Bicyclic RAF InhibitorsPublication Number: JP-2023535595-APriority Date: 2020-07-28



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
| Clinical data | |
|---|---|
| Other names | JZP-815 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2760321-00-2 |
| PubChem CID | 162772363 |
| IUPHAR/BPS | 13233 |
| UNII | P26TTM6U27 |
| KEGG | D13132 |
| Chemical and physical data | |
| Formula | C26H21FN4O3 |
| Molar mass | 456.477 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
{kind=link}
References
- “JZP-815”. PatSnap.
- Riaud M, Maxwell J, Soria-Bretones I, Dankner M, Li M, Rose AA (February 2024). “The role of CRAF in cancer progression: from molecular mechanisms to precision therapies”. Nature Reviews. Cancer. 24 (2): 105–122. doi:10.1038/s41568-023-00650-x. PMID 38195917.
///////////flezurafenib, JZP-815, JZP 815, P26TTM6U27, ANTINEOPLASTIC, CANCER