














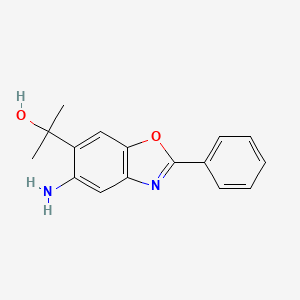
ADX-103
CAS 916056-81-0
Preclinical, Antiinflammatory Ophthalmic Agents, Diabetic Retinopathy,
Agents for Ophthalmic Drugs
MF C16 H16 N2 O2
5-Amino-α,α-dimethyl-2-phenyl-6-benzoxazolemethanol
Aldeyra Therapeutics Inc
ADX-103 , an aldehyde trap being investigated by Aldeyra for the treatment of dry eye syndrome; in May 2018, preclinical data were presented at 2018 ARVO Meeting in Honolulu, HI. Aldeyra, in collaboration with an undisclosed company, is also investigating an anti-inflammatory agent for treating ocular inflammation.
PATENT
WO-2020033344
Novel crystalline forms of a specific benzoxazole and it’s salts, process for their preparation, and compositions comprising them are claimed, useful for treating dry eye, inflammation and diabetes, through action as an aldehyde scavenger.
It has now been found that compounds of the present invention, and compositions thereof, are useful for treating, preventing, and/or reducing a risk of a disease, disorder, or condition in which aldehyde toxicity is implicated in the pathogenesis. In general, salt forms or freebase forms, and pharmaceutically acceptable compositions thereof, are useful for treating or lessening the severity of a variety of diseases or disorders as described in detail herein. Such compounds are represented by the chemical structure below, denoted as compound A:
or a pharmaceutically acceptable salt thereof.
[0008] Compounds of the present invention, and pharmaceutically acceptable compositions thereof, are useful for treating a variety of diseases, disorders or conditions, associated with toxic aldehydes. Such diseases, disorders, or conditions include those described herein.
[0009] Compounds provided by this invention are also useful for the study of certain aldehydes in biology and pathological phenomena.
Scheme 1 – Synthesis of Compound A
Step 1: Synthesis of Compound A2
[00549] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with methanol (10L). Compound A1 (2.0kg) was added, followed by further methanol to rinse (9L). The reaction mixture was warmed to Tjacket=40°C. Once temperature had stabilized, sulfuric acid (220 mL, 0.4eq.) was slowly added. Once addition was complete, agitation was maintained for 30 mins then the vessel was heated to Tjmt=62°C. Reaction progress was
monitored by LC-MS analysis of reaction mixture. The reaction does not go to completion but is deemed complete when no change is apparent in ratio of starting material : product.
[00550] The vessel contents were cooled to Tjmt=24°C and stirred 60 minutes before filtration under vacuum. The filter cake was air dried for 2 hours and the contents then dissolved in ethyl acetate (18L) which was then washed sequentially with saturated sodium bicarbonate (8L), water (8L) and brine (8L) before drying over sodium sulfate, filtration and evaporation in vacuo. Compound A2 (1.5kg, 68.1%) was obtained as a bright orange powder.
Step 2: Synthesis of Compound A3
[00551] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with /V,/V-dimethylformamide (16L). Compound A2 (1.5kg) was added and the brown reaction mixture set to cool to Tint<20oC. Once temperature had stabilized, A-bromosucci ni mi de (l.5kg, 1.1 eq.) was added portion wise, maintaining Tint<27°C. Once addition was complete, the reaction was allowed to stir until starting material content was <1% AUC (250nm) by LCMS analysis.
[00552] A secondary jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with ethyl acetate (16L) and deionized water (22L). The reaction mixture was vacuum transferred into this vessel and held at high agitation for not less than 30 minutes. The aqueous layer was discharged and the organic layer washed with saturated sodium chloride (2 x 8L) then dried over sodium sulfate before evaporation in vacuo to Compound A3 as a deep brown oil (2.lkg, 100.8%), suitable for use in following step without purification.
Step 3: Synthesis of Compound A4
[00553] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with dichloromethane (9L). Compound A3 (2.lkg) was added and the reaction mixture cooled to Tmt<l°C. A solution of Di-/er/-butyl dicarbonate (3.6kg, 2.2 eq.) in dichloromethane (0.5L) was added followed by a solution of A, A-di methyl ami nopyri di ne (92g, 0.1 eq.) in dichloromethane (0.5L). The resultant clear brown solution was stirred for 30 minutes whereupon pyridine (1.3L, 1.7 eq.) was dropwise added, maintaining Tint<5°C. Upon complete addition internal temperature was ramped from Tint=l°C to Tint=20°C over 18 hours.
[00554] The reaction mixture was sequentially washed with saturated sodium chloride (3 x 4.5L), 10 % w/v aqueous citric acid (2 x 4L), saturated sodium bicarbonate (4L), aqueous hydrochloric acid (1M, 4L), saturated sodium bicarbonate (4L) and saturated sodium chloride (4L) then dried over sodium sulfate and evaporated in vacuo with one azeotropic distillation with toluene (2L) to a very dark, heavy tar (3.4kg).
[00555] The isolated tar was mixed with absolute ethanol (3.1L) for 2 days whereupon it was filtered providing light cream colored, granular solids and a black mother liquor. The solids were washed with ice-cold ethanol (3 x 1L) and dried to constant mass. Compound A4 was obtained as off- white granules (1.7 kg, 50.2%).
Step 4: Synthesis of Compound AS
[00556] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with reagent alcohol (6.1 L) and Compound A4 (0.8kg), Tmt<20°C. Iron powder (0.5kg, 5.0 eq.) was added and the suspension stirred vigorously for 30 minutes. Acetic acid (glacial, 1.6L, 15.7 eq.) was added, maintaining Tint<30C.
[00557] Once LCMS confirmed complete consumption of starting material, ethyl acetate (10.2L) and water (10.2L) were added. Sodium bicarbonate (2.3kg, 15.9 eq.) was added portion wise and the layers separated once gas evolution had ceased. The aqueous layer was washed with ethyl acetate until LCMS indicated no further product was being extracted (8 x 2L) and the combined organic layers were sequentially washed with deionized water (6L) then saturated sodium chloride (6L) before drying over magnesium sulfate and evaporation in vacuo. Compound A5 was obtained as a light orange solid (0.7kg, 91.5%).
Step 5: Synthesis of Compound A6
[00558] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with dichloromethane (9L), Compound A5 (0.7kg), and the reaction mixture cooled to Tint 20°C. Benzoyl chloride (0.3L, 1.5 eq.) was added and the reaction stirred 15 minutes. N,N-dimethylaminopyridine (7g, 0.04 eq.) in dichloromethane (0.1L) was added and the reaction stirred 15 minutes. Pyridine (0.5L, 2.5 eq.) was dropwise added, maintaining Tint<20°C. Upon complete addition the reaction was stirred until LCMS indicated consumption of starting material.
[00559] The reaction mixture was washed with deionized water (11L) and the organic layer extracted sequentially with aqueous hydrochloric acid (1M, 3 x 5L), saturated aqueous sodium bicarbonate (11 L), saturated sodium chloride (11 L), dried over magnesium sulfate and evaporated in vacuo. Compound A6 was obtained as a cream colored solid, suitable for use without further purification (0.9kg, 100.7%).
Step 6: Synthesis of Compound A 7
[00560] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with l,2-dimethoxy ethane (16L) and temperature set to Tint=2l°C. Compound A6 (0.9kg) was added and stirred to dissolution. Copper iodide (0.3kg, 1.0 eq.) was added and the mixture stirred 15 minutes. l, lO-phenanthroline (0.3kg, 1.2 eq.) was added and the mixture stirred 15 minutes. Cesium carbonate (l .5kg, 3.0 eq.) was added and the reaction was stirred for 15 minutes. The reaction temperature was ramped to Tint=80-85oC and maintained for 23 hours whereupon it was cooled to Tmt=20°C.
[00561] The reaction mixture was filtered through a celite pad, washing sequentially with deionized water (8L) and ethyl acetate (8L). The organic layer was extracted sequentially with deionized water (2 x 5L), saturated sodium chloride (4L), dried over sodium sulfate and evaporated in vacuo. Compound A7 was obtained as a brown solid, suitable for use without further purification (0.8kg, 104.1%).
Step 7: Synthesis of Compound A8
[00562] A 12L 3 -neck round bottom flask with nitrogen bleed and mechanical stirring was charged with a solution of Compound A7 (0.8kg) in dichloromethane (3.6L) and cooled to Tmt<5°C in an ice bath. Hydrochloric acid in dioxane (4M, 1 2L, 3.1 eq.) was added dropwise with vigorous stirring, maintaining Tmt<25°C. Once addition was complete, the reaction mixture was allowed to stir for 18 hours at Tint=20-25oC.
[00563] The reaction mixture was filtered and the filter cake washed with dichloromethane (2 x 1L) and dried to constant mass. The hydrochloride salt of Compound A8 was isolated as an off-white solid (0.5kg, 88.7%).
Step 8: Synthesis of Compound A
[00564] A 12L 3 -neck round bottom flask with nitrogen bleed and mechanical stirring was charged with a solution of Compound A8 (0.5kg) in tetrahydrofuran (4.8L) and cooled to Tint<-30°C in a dry-ice / acetone bath. Methylmagnesium bromide (3.4M in 2-methyltetrahydrofuran, 2.4L, 5.0eq.) was added slowly, maintaining Tmt<-lO°C. Once addition was complete, the reaction was allowed to warm to room temperature overnight.
[00565] Saturated aqueous ammonium chloride (2L) and ethyl acetate (2L) were added and the reaction mixture stirred for 30 minutes. The aqueous layer was extracted with further ethyl acetate (2 x 2L) and the combined organic layers washed with saturated sodium chloride (2L), dried over sodium sulfate and evaporated in vacuo to a dark heavy oil. The heavy oil was purified by column chromatography on silica gel, eluting with ethyl acetate : heptane 1 : 19 to 1 : 1. Pure Compound A was obtained after evaporation and drying as a brown powder (99.8 g, 23.0%).
Example 1 – Preparation of Free Base Forms A, B and C of Compound A
Compound A
Primary Polymorph Screen
[00566] Based on solubility screen results, a primary polymorph screen using an initial set of 24 solvents, as shown in Table 18, was performed as follows: A) To 24 x 20 mL vials, approximately 50 mg of the received ADX-103 was added; B) The solids were then slurried in 2 mL of the solvents and left placed in an incubator/shaker to temperature cycle between ambient and 40 °C in 4 hour cycles; C) After 72 hours temperature cycling, the mother liquors were removed from the vials and split evenly between 4 x 2 mL vials. The vials were then split between evaporation, crash cooling to 2 °C and -18 °C and anti-solvent addition; and D) Any solids
recovered were analysed by XRPD, any new patterns identified were also analysed by TG/DTA and PLM.
Table 18. Solvents Selected for Initial Primary Polymorph Screen
PATENT
WO2018039197 , as compound I-8.
PATENT
WO 2006127945
WO 2011072141
WO 2014116593
US 20150344447
WO 2020028820
////////////ADX-103, Preclinical, Antiinflammatory, Ophthalmic Agents, Diabetic Retinopathy, Aldeyra Therapeutics Inc,
CC(C)(O)c1cc2oc(nc2cc1N)c3ccccc3