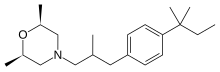
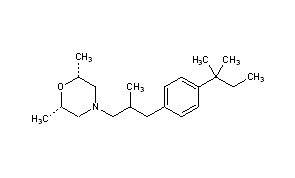
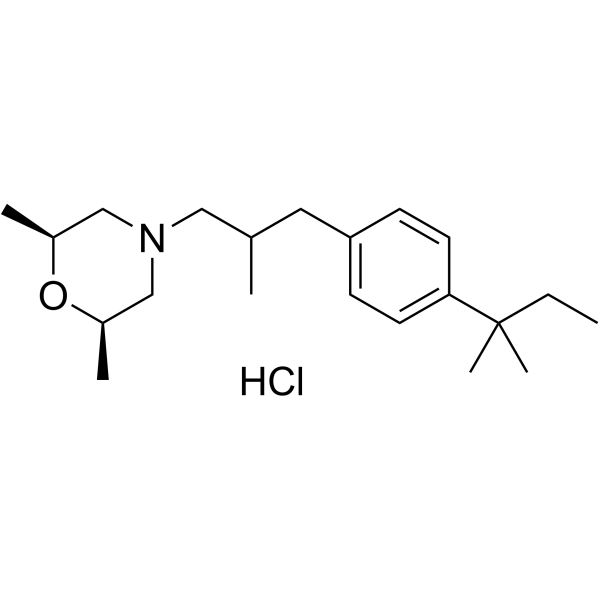
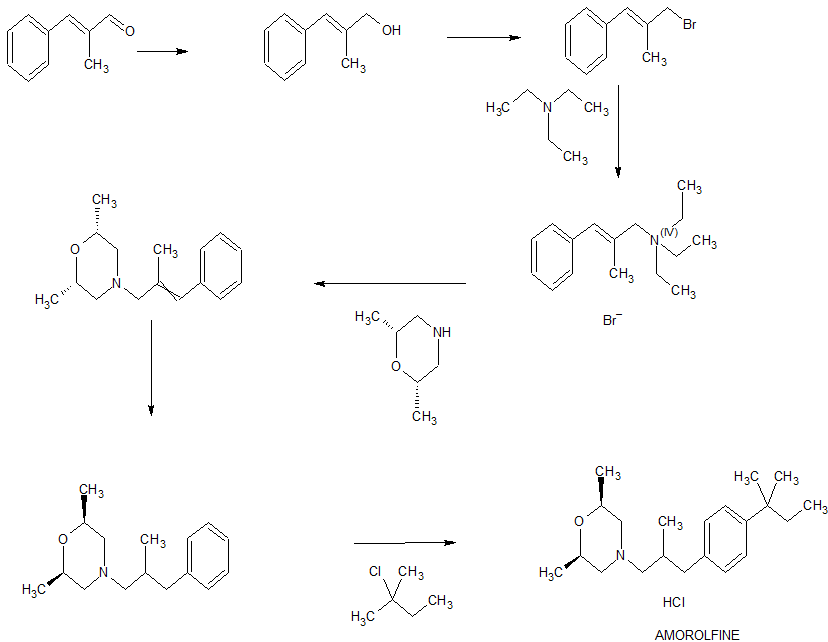
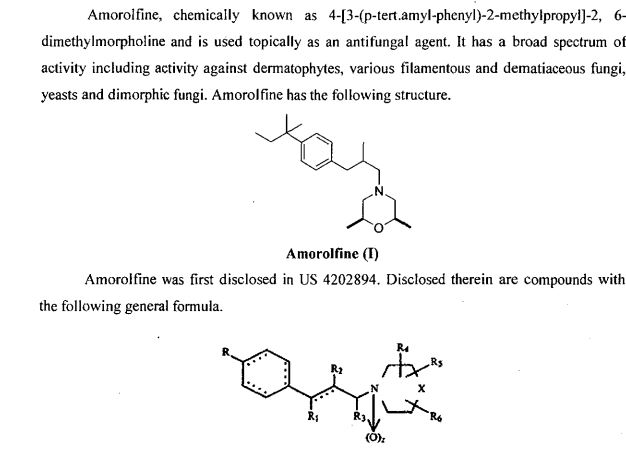
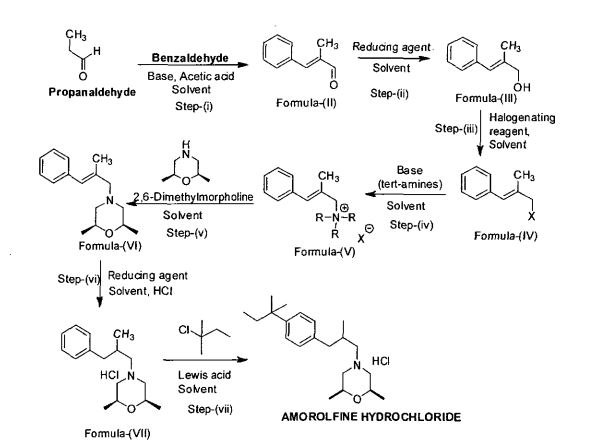
In a 10L clean reaction kettle, add 2600 mL of acetic anhydride, 5200 mL of glacial acetic acid, 350 g of sodium periodate, break 1236 g, cool to 5 ° C, add 810 mL of sulfuric acid, control the dropwise addition within 1 hour, and then add 1130 g of t-amyl. The benzene was stirred at room temperature for more than 16 hours, and the reaction of the raw materials was confirmed by thin layer chromatography. The reaction mixture was poured into a mixture of 8 L of water and 4 L of dichloromethane, and the mixture was separated. The organic layer was washed with 4L of 25% aqueous sodium sulfite, and the organic layer was dried over anhydrous sodium sulfate. It was 4-iodo-t-amylbenzene 2013 g, yield: 96%, and the GC purity was 94.2%. NMR spectral data: (400 MHz, CDC1 3 ): 0.73 (3H, t, J = 7.4 Hz), 1.31 (6H, s), 1.67 (2H, q, J – 7.4 Hz), 7.13 (2H, d, J = 8.56 Hz), 7.66 (2H, d, J = 8.56 Hz) 0 Example 2
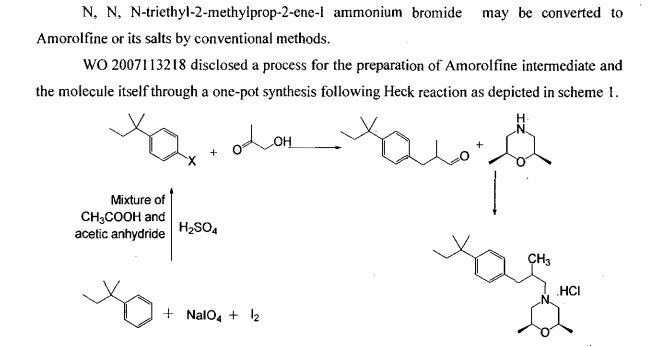
2 kg of 4-iodo-t-amylbenzene prepared according to the method of Example 1 and 6 L of N-methylpyrrolidone were added to a 10 L clean reaction vessel, and the mixture was stirred under nitrogen, stirring was carried out, and 300 g of palladium acetate and 1.7 kg of sodium hydrogencarbonate were added. Finally, 2.5 kg of 2-mercaptopropanol was added, the temperature was raised to 105 C, and the GC content of 4-iodo-t-amylbenzene was measured to monitor the progress of the reaction, and the reaction was completed for 2 hours. Cool to room temperature, filter, concentrate the filtrate, add the residue to 12 L of ethyl acetate, wash with 20 L of water, rectify the organic phase, collect 125-128 ° C fraction (vacuum degree ≤ -0.099)\3⁄4^), and obtain 3- Tert-amylphenyl-2-mercaptopropanal L41 kg, yield: 88.6%, GC purity: 93.5%. NMR spectral data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J = 7.45 Hz), 1.11 (3H, d, J = 6.87 Hz), 1.29 (6H, s), 1.65 (2H, q, J =7.43 Hz), 2.60 (13⁄4 dd, J=13.52 Hz), 2.69 (1H, J=7.06 Hz), 3.08 (1H, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz), 7.27 (2H, d, J = 8.27 Hz), 9,75 (1H, s).
The above 3-tert-pentylphenyl-2-methylpropanal lkg, 5 L of ethyl acetate was added to a 10 L reactor, protected with nitrogen, cooled to 10 ° C, and 600 g of 2,6-dimethylmorpholine was added dropwise. , add about 30 minutes. Then, 300 mL of glacial acetic acid was added dropwise, the temperature was kept at 15 C, the addition was completed, and the temperature was raised to 18 ° C for 30 minutes. After cooling to 10 Torr, 1,3 kg of sodium triacetoxyborohydride was added. After the addition, the temperature was maintained at 18 ° C, and the GC content of 3-tert-amylphenyl-2-methylpropanal was detected to monitor the progress of the reaction. Ended in 2 hours. After cooling to 10 ° C or lower, the pH was adjusted to 10 with a sodium hydroxide solution, and the layers were allowed to stand, and the organic layer was washed with 4 L of water. The organic phase was added with concentrated hydrochloric acid, adjusted to pH 2, filtered, and the filter cake was dried under reduced pressure at 65 V for 14 hours to obtain 1.59 kg of amorolamine hydrochloride, yield: 85.6%, HPLC purity: 99.6%. R spectrum data: 3⁄4 NMR (400MHz, CD 3 OD) 5: 0.64 (3H, t, J=7, 2Hz), 1.03 (3H, d, J=6.8Hz), 1.15(6H, d, J=6 , 0 Hz), 1.25 (63⁄4 s), 1.64 (2H, m, J = 7.6 Hz), 2.34 (1H, d, J = 6.8 Hz), 2.48 (23⁄4 d, J = 6.8 Hz), 2.75 (2H, d , J=6.0Hz), 3.1(2H, d, J=8.8Hz) 5 3.4(2H, d, J=11.2Hz), 3,9(2H, m), 7.16(2H, dd, J=8.4Hz ), 7.27 (2H, dd, J = 8.4 Hz). Example 3 In a 10 L clean reaction kettle, 2 kg of 4-substituted tert-amylbenzene prepared according to the method of Example 1 and 6 L of N-mercaptopyrrolidone were protected by nitrogen, stirring was started, and 150 g of palladium acetate and 2.5 kg of dipotassium hydrogen phosphate were added. Finally, 1.8 kg of 2-methylallyl alcohol was added, and the temperature was raised to 130. C reaction, the GC content of 4-deuterated tert-amylbenzene was measured to control the progress of the reaction, and the reaction was completed for 10 hours. Cool to room temperature, filter, concentrate the filtrate, add the residue to 12 L of ethyl acetate, dissolve 20 L of water, concentrate the organic phase, recover ethyl acetate, and add the residue to 10 L of saturated sodium hydrogen sulfite solution at room temperature to precipitate solid. The mixture was stirred for 6 hours, filtered, and filtered, washed with EtOAc EtOAc EtOAc EtOAc. The filtrate was concentrated to dry ethyl acetate to give 1. <RTI ID=0.0>#</RTI><RTIgt;</RTI><RTIgt;</RTI><RTIgt; -NMR spectral data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J-7.45 Hz), 1.11 (3H, d, J-6.87 Hz), 1.29 (6H, s), 1.65 (2H, q, J=7.43 Hz), 2.60 (1H, dd, J=13.52 Hz), 2.69 (1H, J=7.06 Hz), 3.08 (1H, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz) ), 7.27 (2H, d, J = 8.27 Hz), 9.75 (1H, s).
Add 1 kg of the above 3-tert-pentylphenyl-2-methylpropanal, 5 L of ethyl acetate in a 10 L reactor, protect with nitrogen, cool to 10 C, and add 1.2 kg of 2,6-dimethylmorpholine dropwise. , 40 minutes added. Then, 780 mL of glacial acetic acid was added dropwise, the temperature was kept at 15 ° C, the addition was completed, and the temperature was raised to 20 ° C for 60 minutes. After cooling to 10 ° C, 2.3 kg of sodium triacetoxyborohydride was added. After the addition, the temperature was checked at 25 ° C, and the GC content of 3-tert-amylpyridyl-2-methylpropanal was detected to monitor the progress of the reaction. The reaction was completed in 2 hours. Cool to below 10 ,, adjust the pH to 11 with sodium hydroxide solution, let stand for stratification, wash the organic layer with 4 L of water, add concentrated hydrochloric acid to the organic phase, adjust pH to 2, filter, filter cake at 70 ° C decompression After drying for 14 hours, 1.75 kg of amorolfine hydrochloride was obtained, yield: 84.6%, HPLC purity: 99.7%. R spectrum data: 3⁄4 NMR (400MHz, CD 3 OD) 5: 0.64 (3H, t, J = 7.2Hz), 1.03 (3H, d, J = 6.8Hz), L15(6H, d, J=6.0Hz ), 1.25(6H, s), L64(2H 5 m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75(2H, d , J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H 5 d, J=11.2Hz), 3·9(2Η, m), 7.16(2H, dd, J=8.4Hz ), 7.27 (2H ; dd, J = 8.4 Hz). Example 4
In a 10 L clean reaction kettle, 2 kg of 4-iodo-t-amylbenzene prepared according to the method of Example 1, 2 N of N-methylpyrrolidone, protected by nitrogen, stirring was started, and palladium nitrate 6 g, acetic acid was added. Sodium 627 g, and finally 592 g of 2-methylallyl alcohol was added thereto, and the temperature was raised to 140 ° C to carry out a reaction. The GC content of 4-deactivated t-amylbenzene was examined to monitor the progress of the reaction, and the reaction was terminated for 24 hours. Cool to room temperature, filter, concentrate the filtrate, add the residue to 8 L of ethyl acetate, dissolve in 16 L of water, rectify the organic phase, collect 125-128 C fraction (vacuum degree ≤ -0.0991 ^ & ) to give 3-tert-pentylphenyl 2-mercaptopropanal 1.37 kg, yield: 86%, GC purity: 93.0%. MR spectrum data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J = 7.45 Hz), 1 , 11 (3H, d, J = 6.87 Hz), 1.29 (6H, s), 1.65 (2H, q , 3=1 A3 Hz), 2.60 (IH, dd, J=13.52 Hz), 2.69 (IH, J=7.06 Hz), 3.08 (IH, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz), 7.27 (2H, d, J = 8.27 Hz), 9.75 (IH, s).
The above 3-tert-pentylphenyl-2-mercaptopropanal lkg, 5 L of dichloromethane was added to a 10 L reactor, protected with nitrogen, cooled to 10 ° C, and 1.6 kg of 2,6-dimethyl was added dropwise. Morpholine, added in 45 minutes. Then, 300 mL of glacial acetic acid was added dropwise, the temperature was kept at 15 ° C, the addition was completed, and the temperature was raised to 23 Torr for 60 minutes. After cooling to 10 ° C, 1.6 kg of sodium triacetoxyborohydride was added. After the addition, the temperature was checked at 23 ° C, and the GC content of 3-tert-pentylphenyl-2-methylpropanal was detected to monitor the progress of the reaction. The end of the hour. Cool to below 10 °C, adjust the pH to 10 with sodium hydroxide solution, let stand for layering, wash the organic layer with 4L water, add concentrated hydrochloric acid to the organic phase, adjust the pH to 1, filter, filter cake at 70 °C After drying under reduced pressure for 14 hours, 1.59 kg of amorolamine hydrochloride was obtained, yield: 83.6%, HPLC purity: 99.6%. iH-NMR spectral data: ! H NM (400 MHz, CD 3 OD) 5: 0.64 (3H, t, J = 7.2 Hz), 1.03 (3H, d, J = 6.8 Hz), 1.15 (6H, d, J= 6.0Hz), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75(2H , d, J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J=11.2Hz), 3.9(2H, m), 7.16(2H, dd, J=8.4Hz ), 7.27 (2H, dd, J = 8.4 Hz). Example 5
2 kg of 4-iodo-t-amylbenzene prepared according to the method of Example 1 and 4 L of N-methylpyrrolidone were added to a 10 L clean reaction vessel, and the mixture was stirred under nitrogen, stirring was carried out, 30 g of palladium chloride and 750 g of sodium hydrogencarbonate were added. Finally, 1.3 kg of 2-methylallyl alcohol was added, and the mixture was heated to 120 ° C to measure the GC content of 4-iodo-t-amylbenzene to control the progress of the reaction, and the reaction was completed for 13 hours. It was cooled to room temperature, filtered, and the filtrate was concentrated. The residue was dissolved in 8 L of chloroform, washed with 16 L of water, and the organic phase was concentrated. The ethyl acetate was recovered. The residue was added dropwise to 10 L of saturated sodium hydrogensulfite solution at room temperature to precipitate a solid. Hour, filter, filter cake washed with 5 L of ethyl acetate, solid dispersed in 3 L 3 mol / liter The mixture was stirred at room temperature for 5 hours, and the reaction mixture was dried over EtOAcjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjj Yield: 91,7%, GC purity: 98.8%. – Spectrum data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J=7.45 Hz), 1.11 (3H, d, J-6.87 Hz), 1.29 (6H, s), 1.65 (2H, q, J=7.43 Hz), 2.60 (IH, dd, J=13.52 Hz), 2.69 (IH, J=7.06 Hz), 3.08 (IH, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz) ), 7.27 (2H, d, J = 8.27 Hz), 9.75 (IH, s).
Add 1 kg of the above 3-tert-pentylphenyl-2-methylpropanal, 5 L of absolute ethanol in a 10 L reactor, protect with nitrogen, cool to 10 ° C, and add 600 g of 2,6-dimercaptomorpholine. , added in 30 minutes. Then, 500 mL of glacial acetic acid was added dropwise, the temperature was kept at 15 ° C, the addition was completed, and the temperature was raised to 23 ° C for 60 minutes. After cooling to 10 ° C, 1.2 kg of sodium triacetoxyborohydride was added. After the addition, the temperature was monitored at 10 Torr, and the GC content of 3-tert-pentylphenyl-2-nonylpropionaldehyde was detected to monitor the progress of the reaction. The end of the hour. 10. Under C, adjust the pH value to 11 with sodium hydroxide solution, add 3 L of dichloromethane, let stand for layering, wash the organic layer with 4 L of water, add concentrated hydrochloric acid to the organic phase, adjust pH to 2, filter, filter cake at 7 CTC minus After drying for 14 hours, 1.45% of amorolfine hydrochloride was obtained, yield: 87.0%, HPLC purity: 99.7% – NMR spectral data: J H NMR (400 MHz 5 CD 3 OD) 6: 0.64 (3H, t, J= 7,2Hz), 1.03(3H, d, J=6.8Hz), 1.15(6H, d, J=6.0Hz), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34( 1H ? d, J = 6.8 Hz), 2.48 (2H, d, J = 6.8 Hz), 2.75 (23⁄4 d, J = 6.0 Hz), 3.1 (2H, d, J = 8.8 Hz), 3.4 (2H, d , J = 11.2 Hz) 5 3.9 (2H, m), 7.16 (2H, dd, J = 8.4 Hz), 7.27 (2H, dd, J = 8.4 Hz). Example 6
2 kg of 4-iodo-t-amylbenzene prepared in accordance with the method of Example 1 and 4 L of N-methylpyrrolidone were added to a 10 L clean reaction vessel. The mixture was stirred under nitrogen, stirring was started, 10 g of palladium acetate was added, and 800 g of carbonic acid was added. 1.1 kg of 2-mercaptopropanol was heated to 80 ° C, and the GC content of 4-deactivated t-amylbenzene was measured to control the progress of the reaction, and the reaction was terminated for 24 hours. Cool to room temperature, filter, concentrate the filtrate, add 8 L of chloroform to dissolve, 16 L of water, rectify the organic phase, collect 125-128 ° C 真空 (vacuum degree ≤ -0.099 ^ ^ & ), to obtain 3-tert-amylbenzene Base-2-mercaptopropanal 1.42 kg, yield: 89.2%, GC purity: 92.5%. ^- MR Spectral Data: (400 MHz, CDC1 3 ): 0.69 (33⁄4 t, J=7.45 Hz), 1.11 (3H, d, J=6.87 Hz), 1.29 (6H, s), 1.65 (2H, q, J=7.43 Hz), 2.60 (IH, dd, J=13.52 Hz), 2.69 (IH, J=7.06 Hz), 3.08 (IH, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz), 7.27 (2H, d, J = 8.27 Hz), 9.75 (1H, s).
The above 3-tert-pentylphenyl-2-methylpropanal lkg, 5 L of decyl alcohol was added to a 10 L reactor, protected with nitrogen, cooled to 10 C, and 600 g of 2,6-dimethylmorpholine was added dropwise for 30 minutes. Plus finished. Then, 500 mL of water acetic acid was added dropwise, the temperature was kept at 10 ° C, the addition was completed, and the temperature was raised to 20 ° C for 60 minutes. After cooling to 10 C, 1.2 kg of sodium triacetoxyborohydride was added. After the addition, the temperature was maintained at 23 ° C, and the GC content of 3-tert-pentylphenyl-2-methylpropanal was detected to monitor the progress of the reaction. End of 2 hours. Cool to 10 ° C, adjust the pH to 10 with sodium hydroxide solution, add 3 L of dichloromethane, let stand for layering, wash the organic layer with 4 L of water, add concentrated hydrochloric acid to the organic phase, adjust pH to 1.5, filter, filter The cake was dried under reduced pressure at 65 C for 15 hours to obtain 1.46 kg of amorolfine hydrochloride, yield: 90.1%, HPLC purity: 99,8%. ^-NMR spectral data: l R NMR (400 MHz, CD 3 OD) 5: 0.64 (3H, t, J = 7.2 Hz), 1.03 (3H, d, J = 6.8 Hz), U5 (6H, d, J = 6.0Hz), 1.25(6H, s), 1.64(23⁄4 m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75(2H, d, J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J=l 1.2Hz), 3.9(2H, m), 7.16(2H, dd, J=8.4Hz ), 7.27 (2H, dd, J = 8.4 Hz). Example 7
2 kg of 4-iodo-t-amylbenzene prepared according to the method of Example 1 and 6 L of N-decylpyrrolidone were added to a 10 L clean reaction kettle, protected by nitrogen, stirring was started, and 75 g of palladium acetate and 2.0 kg of disodium hydrogen phosphate were added. Finally, 780 g of 2-methylallyl alcohol was added, and the temperature was raised to 125 Torr. The GC content of 4-iodo-t-amylbenzene was measured to control the progress of the reaction, and the reaction was terminated for 8 hours. The mixture was cooled to room temperature, filtered, and the filtrate was concentrated. The residue was evaporated, evaporated, evaporated, evaporated, evaporated. The solid was precipitated, stirred for 6 hours, filtered, and the filter cake was washed with 5 L of ethyl acetate. The solid was dispersed in 10 L 2 mol/L hydrochloric acid, stirred at room temperature for 5 hours, and the reaction mixture was extracted with 10 L of ethyl acetate. The mixture was dried, filtered, and the filtrate was evaporated to ethyl acetate to ethylamine (ethyldiethyldithioacetate). 3⁄4-NMR spectral data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J = 7.45 Hz), 1.11 (3H, d, J = 6.87 Hz), 1.29 (6H, s), 1.65 (2H, q , J=7.43 Hz), 2.60 (1H, dd, J=13.52 Hz), 2.69 (1H, J=7.06 Hz), 3.08 (1H, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz), 7.27 (2H, d, J = 8.27 Hz), 9.75 (1H, s).
Add the above 3-tert-pentylphenyl-2-mercaptopropanal lkg, 5L hydrazine, in a 10L reactor Under nitrogen atmosphere, cooled to 10 Torr, 700 g of 2,6-dimercaptomorpholine was added dropwise, then 280 mL of glacial acetic acid was added, the temperature was maintained at 15 C, and then the temperature was raised to 23 ° C for 60 minutes. After cooling to 10 ° C, 1.0 kg of sodium triacetoxyborohydride was added, and 20 was added. The temperature was maintained under C, and the GC content of 3-tert-amylphenyl-2-methylpropanal was examined to monitor the progress of the reaction, and the reaction was completed for 3 hours. Cool to below 10 ° C, adjust the pH to 11 with sodium hydroxide solution, let stand for layering, wash the organic layer with 4 L of water, add concentrated hydrochloric acid to the organic phase, adjust the pH to 1, filter, filter cake at 70 ° C After drying under reduced pressure for 14 hours, 1.59 kg of amorolamine hydrochloride was obtained, yield: 83.8%, HPLC purity: 99.6%. ^-NMR spectral data: 3⁄4 NMR (400MHz, CD 3 OD) 5: 0.64 (3H, t, J- 7.2 Hz), 1.03 (3H, d, J = 6.8 Hz), 1.15 (6H ; d, J = 6.0 Hz), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz) } 2.75(2H, d, J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J=11.2Hz), 3.9(2H, m), 7.16(2H, dd, J=8.4Hz) , 7.27 (2H, dd, J = 8.4 Hz). Example 8
3-tert-pentylphenyl-2-mercaptopropanol lkg, 5 L of dichloromethane prepared by the method of Example 5 was added to a 10 L reactor, and was purged with nitrogen and cooled to 10. C, 1000 g of 2,6-dimethylmorpholine was added dropwise, then 400 mL of water acetic acid was added, the temperature was maintained at 15 ° C, and then the temperature was raised to 20 ° C for 60 minutes. After cooling to 0 C, 1.5 kg of sodium triacetoxyborohydride was added, and 6 C was added after the addition, and the GC content of 3-tert-pentylphenyl-2-mercaptopropanal was detected to monitor the progress of the reaction for 5 hours. End. Adjust the pH to 10 with sodium hydroxide solution at 6 °C, let stand for layering, wash the organic layer with 4L of water, add concentrated hydrochloric acid to the organic phase, adjust the pH to 2, filter, filter cake and dry at 65 Ό for 14 hours under reduced pressure. , Amofufen hydrochloride 1.48kg, yield: 91.2%, HPLC purity: 99.7%. ^- MR spectral data: NMR (400MHz, CD 3 OD) 5: 0·64 (3Η, ΐ, J=7, 2Hz), 1.03(3Η, d, J=6.8Hz), 1.15(6H, d, J =6.0Hz), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75( 2H, d, J=6.0Hz), 3,1(2H, d, J=8.8Hz), 3.4(2H, d, J=11.2Hz), 3.9(23⁄4 m), 7.16(2H, dd, J- 8.4 Hz), 7.27 (2H, dd, J = 8.4 Hz). Example 9
Add 3-tert-pentylphenyl-2-mercaptopropanol lkg prepared in the same manner as in Example 2, 4 L of tetrahydrofuran, protect with nitrogen, cool to 10 ° C, add 820 g of 2,6-two Mercaptomorpholine, Then, 380 mL of glacial acetic acid was added, the temperature was maintained at 15 ° C, and then kept at room temperature for 60 minutes. After cooling to 10 ° C, 1.8 kg of sodium triacetoxyborohydride was added, and after 10 liters of the addition, the GC content of 3-tert-amylphenyl-2-nonylpropionaldehyde was detected to monitor the progress of the reaction for 5 hours. End. The pH was adjusted to 10 with sodium hydroxide solution at 10 ° C, and the layers were allowed to stand. The organic layer was washed with 4 L of water, and the organic phase was added with concentrated hydrochloric acid, adjusted to pH 2, filtered, and the filter cake was dried under reduced pressure at 65 Torr for 14 hours. , amlofol hydrochloride 1.41 kg, yield: 87.1%, HPLC purity: 99.8%. NMR spectral data: J H NMR (400 MHz, CD 3 OD) 5: 0.64 (3H, t, J- 7.2 Hz), L03 (3H, d, J = 6.8 Hz), 1.15 (6H, d, J = 6.0 Hz) ), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34(1H, d, J-6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75(2H, d , J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J=11.2Hz), 3.9(2H, m), 7.16(2H, dd, J=8.4Hz), 7.27 (2H, dd, J = 8.4 Hz). Comparative example 1
In a 1000 mL four-necked flask, 137 g of 4-deuterated tert-amylbenzene prepared according to the method of Example 1, 1.12 g of palladium acetate, 50.4 g of sodium hydrogencarbonate, N,N-dimethylformamide 500 mL, nitrogen gas, added 54 g of 2-mercaptopropanol, warmed to 10 (TC for 10 hours, cooled to room temperature, filtered, filter cake washed with hydrazine, hydrazine-dimethylformamide 300 mL, combined filtrate, poured into 2000 mL of saturated brine and 1000 mL The mixture was extracted with ethyl acetate, and the organic phase was washed with water, dried over anhydrous magnesium sulfate, filtered, and concentrated, dried, and evaporated, and the residue was distilled in vacuo to collect fractions of 125-128 ° C (vacuum degree <-0.099 MPa) to obtain 3-un Amyl phenyl-2-mercaptopropanal 84 g, Yield: 77%, GC purity: 88.0% – R spectrum data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J = 7.45 Hz) , 1.11 (3H : d, J=6.87 Hz), 1.29 (6H, s), 1.65 (2H, q, J=7.43 Hz), 2,60 (1H, dd, J=13.52 Hz), 2.69 (1H, J-7.06 Hz), 3.08 (1H, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz), 7.27 (2H, d, J = 8.27 Hz), 9.75 (1H, s).
109 g of the above 3-tert-amylphenyl-2-mercaptopropanal and 500 mL of ethanol were placed in a 1000 mL four-necked flask, cooled to 0 ° C, and 30 mL of glacial acetic acid and 69 g of 2,6-dimethylmorpholine were added. Stir at room temperature for 30 minutes, cool to -15 ° C, add 15.93 g of sodium borohydride in 1 hour. After the addition, warm to 0 C for 2 hours, adjust the pH to 12 with 25% sodium hydroxide solution. The mixture was extracted with 2000 mL of saturated brine and 1000 mL of ethyl acetate. The organic phase was washed with water and concentrated to dryness. The obtained residue was added to 500 mL of isopropyl ether, hydrogen chloride gas to pH 2, stirred at room temperature for 2 hours, filtered, and washed with isopropyl ether. , the filter cake is dried under reduced pressure at 70 ° C for 14 hours to obtain hydrochloric acid. Morofen 119 g, yield: 67%, HPLC purity: 97.1%. 3⁄4-NMR spectral data: ‘H NMR (400 MHz, CD 3 OD) 5: 0, 64 (3H, t, J = 7, 2 Hz), 1.03 (3H, d, J = 6.8 Hz), 1.15 (6H, d , J=6.0Hz), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75(2H, d, J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J=11.2Hz), 3,9(2H, m), 7.16(2H, dd , J = 8.4 Hz), 7.27 (2H, dd, J = 8.4 Hz). Comparative example 2
109 g of 3-tert-pentylphenyl-2-methylpropanal prepared according to the method of Comparative Example 1 and 500 mL of methanol were added to a 1000 mL four-necked flask, cooled to 0 ° C, and 30 mL of glacial acetic acid and 69 g of 2, 6 were added. – dimethylmorpholine, stirred at room temperature for 30 minutes, cooled to -15 ° C, replaced with nitrogen, added 5 g of 0% palladium on carbon, passed through hydrogen, reduced at 40 ° C, 4 atm, until the hydrogen pressure did not decrease, The reaction is complete. Cool to room temperature, replace with nitrogen, filter, adjust the pH of the filtrate with 25% sodium hydroxide solution, add 2000 mL of saturated brine and 1000 mL of ethyl acetate for extraction, wash the organic phase, concentrate and dry, add the residue to 500 mL Isopropyl ether, hydrogen chloride gas to pH 2, stirred at room temperature for 2 hours, filtered, washed with isopropyl ether, and the filter cake was dried under reduced pressure at 70 ° C for 14 hours to obtain amolofol hydrochloride 113 g, yield: 64%. HPLC purity: 97.8%. NMR spectral data: 3⁄4 NMR (400MHz, CD 3 OD) 5: 0.64 (3H, t, J = 7.2 Hz), 1.03 (3H, d, J = 6.8 Hz), 1.15 (6H, d, J = 6.0 Hz) , 1.25(6H, s), 1.64(2H, m, J-7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz) ? 2.75(2H, d, J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J-11.2Hz), 3.9(2H, m), 7.16(2H, dd, J=8.4Hz), 7.27 (2H, dd, J=8, 4Hz).