














 |
||
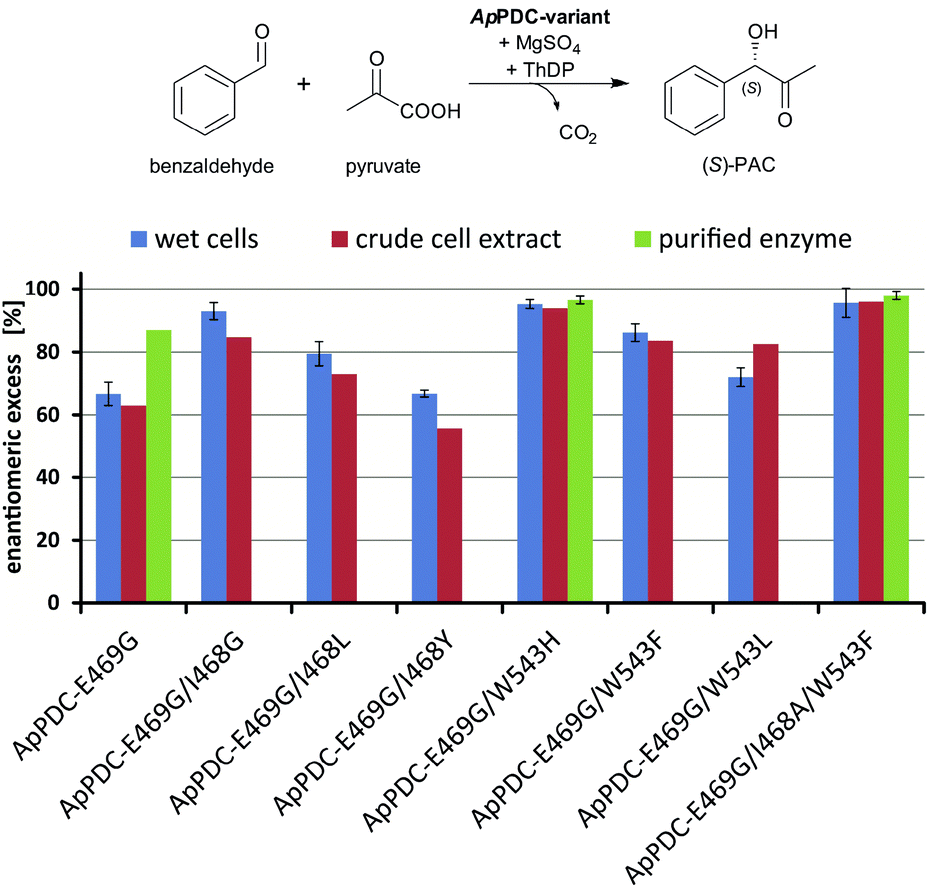
| Fig. 3 Stereoselectivities of the new ApPDC-variants for the synthesis of (S)-PAC. The different variants were tested as wet cells, crude cell extracts, and purified enzymes. Reaction conditions: wet cells – 20 mM benzaldehyde; 200 mM pyruvate; 50 mM KPi-buffer (pH 6.5), 2.5 mM MgSO4; 0.1 mM ThDP; 20 °C; 800 rpm, 800 μL reaction volume in 1.5 mL closed glass vials, whole cell catalyst concentration of 50 mg mL−1. Crude cell extract – 20 mM benzaldehyde; 200 mM pyruvate; 50 mM KPi-buffer (pH 6.5), 2.5 mM MgSO4; 0.1 mM ThDP; 20 °C; 800 rpm, 500 μL reaction volume in a 96-well sheet; see ESI chapter 2.1.4–2.1.5† for the catalyst concentration. Purified enzyme – 40 mM benzaldehyde; 200 mM pyruvate; 50 mM KPi-buffer with three different pH values, 2.5 mM MgSO4; 0.1 mM ThDP; 22 °C; 800 rpm, 800 μL reaction volume in 1.5 mL closed glass vials; protein concentration of 1 mg mL−1. | ||
Asymmetric synthesis of (S)-phenylacetylcarbinol – closing a gap in C–C bond formation
E-mail: do.rother@fz-juelich.de
DOI: 10.1039/C6GC01803C
(S)-Phenylacetylcarbinol [(S)-PAC] and its derivatives are valuable intermediates for the synthesis of various active pharmaceutical ingredients (APIs), but their selective synthesis is challenging. As no highly selective enzymes or chemical catalysts were available, we used semi-rational enzyme engineering to tailor a potent biocatalyst to be >97% stereoselective for the synthesis of (S)-PAC. By optimizing the reaction and process used, industrially relevant product concentrations of >48 g L−1 (up to 320 mM) were achieved. In addition, the best enzyme variant gave access to a broad range of ring-substituted (S)-PAC derivatives with high stereoselectivity, especially for meta-substituted products.
 |
||
| Fig. 2 Schematic representation of the active site of ApPDC. The legends explain the effect of different amino acid residues on the preferred orientation of the ThDP-bound donor substrate acetaldehyde, derived from pyruvate after decarboxylation (green rectangle) and the aromatic acceptor aldehyde (blue hexagon). The relative orientation of both substrates to each other defines the stereoselectivity of the product. (The figures refer to the stereoselectivities achieved with purified enzyme.) | ||
///////////////Asymmetric synthesis, (S)-phenylacetylcarbinol, C–C bond formation