














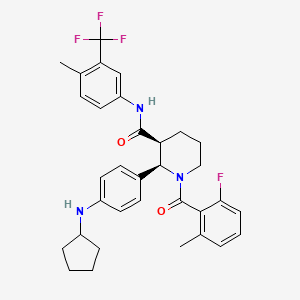

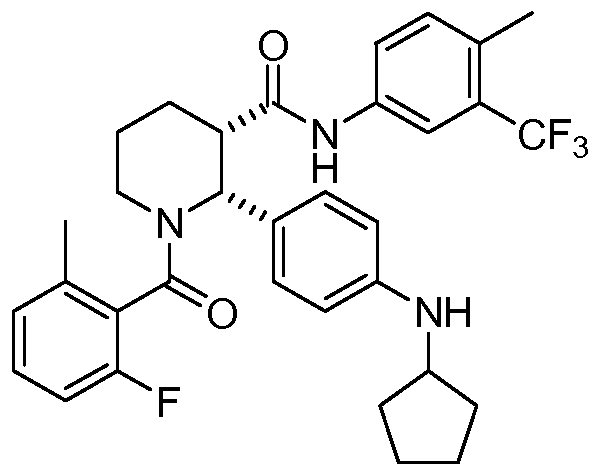
Avacopan
アバコパン
| Formula |
C33H35F4N3O2
|
|---|---|
| CAS | 1346623-17-3 |
| Mol weight |
581.6435
|
(2R,3S)-2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methylbenzoyl)-N-[4-methyl-3-(trifluoromethyl)phenyl]piperidine-3-carboxamide
(2R,3S)-2-(4-Cyclopentylaminophenyl)-l-(2-fluoro-6-methylbenzoyl)piperidine-3- carboxylic acid (4-methyl-3-trifluoromethylphenyl)amide
3-Piperidinecarboxamide, 2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methylbenzoyl)-N-[4-methyl-3-(trifluoromethyl)phenyl]-, (2R,3S)-
- (2R,3S)-2-[4-(Cyclopentylamino)phenyl]-1-(2-fluoro-6-methylbenzoyl)-N-[4-methyl-3-(trifluoromethyl)phenyl]-3-piperidinecarboxamide
- (2R,3S)-2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methylbenzoyl)-N-[4-methyl3-(trifluoromethyl)phenyl]piperidine-3-carboxamide
APPROVED PMDA JAPAN 2021/9/27, Tavneos

Anti-inflammatory, Complement C5a receptor antagonist
Treatment of anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis
Avacopan
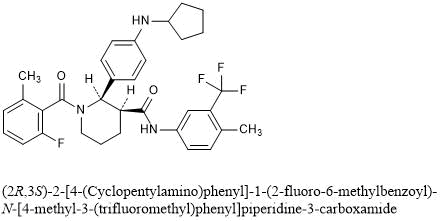
(2R,3S)-2-[4-(Cyclopentylamino)phenyl]-1-(2-fluoro-6-methylbenzoyl)-N-[4-methyl-3-(trifluoromethyl)phenyl]piperidine-3-carboxamide
C33H35F4N3O2 : 581.64
[1346623-17-3]
CCX 168
Avacopan wasunder investigation in clinical trial NCT02994927 (A Phase 3 Clinical Trial of CCX168 (Avacopan) in Patients With ANCA-Associated Vasculitis).
VFMCRP announces approval for TAVNEOS® (avacopan) for the treatment of ANCA-associated vasculitis in Japan
- First orally administered therapy for the treatment of two types of ANCA-associated vasculitis approved in Japan
- Partner Kissei to market TAVNEOS® in Japan, with launch expected as soon as possible following National Health Insurance (NHI) price listing
ST. GALLEN, Switzerland–(BUSINESS WIRE)–Vifor Fresenius Medical Care Renal Pharma (VFMCRP) today announced that Japan’s Ministry of Health and Labor Welfare (MHLW) has granted its partner, Kissei Pharmaceutical Co., Ltd., marketing authorization approval for TAVNEOS® for the treatment of patients with granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA), the two main types of ANCA-associated vasculitis, a rare and severe autoimmune renal disease with high unmet medical need.
“We are delighted that TAVNEOS® has been approved in Japan, the first market worldwide, and congratulate our partner Kissei for this significant milestone”
“We are delighted that TAVNEOS® has been approved in Japan, the first market worldwide, and congratulate our partner Kissei for this significant milestone,” said Abbas Hussain, CEO of Vifor Pharma Group. “ANCA-associated vasculitis is officially designated an intractable disease in Japan, indicating a rare disease without any effective treatment but for which long-term treatment is required. There is significant unmet medical need of over 10,000 patients in Japan, and we believe in the potential of TAVNEOS® for treating it. We are confident that Kissei will fully focus on bringing this breakthrough treatment to this patient population, helping them lead better, healthier lives.”
The approval is based on the marketing authorization application filing by Kissei which was supported by positive clinical data from the pivotal phase-III trial ADVOCATE in a total of 331 patients with MPA and GPA in 18 countries and regions, including Japan. TAVNEOS® demonstrated superiority over standard of care at week 52 based on Birmingham Vasculitis Activity Score (BVAS).
VFMCRP holds the rights to commercialize TAVNEOS® outside the U.S.. In June 2017, VFMCRP granted Kissei the exclusive right to develop and commercialize TAVNEOS® in Japan. Kissei expects to begin to market TAVNEOS® as soon as possible following NHI price listing. Outside Japan, TAVNEOS is currently in regulatory review with various agencies, including the U.S. Food and Drug Administration and the European Medicines Agency.
About Vifor Pharma Group
Vifor Pharma Group is a global pharmaceuticals company. It aims to become the global leader in iron deficiency, nephrology and cardio-renal therapies. The company is a partner of choice for pharmaceuticals and innovative patient-focused solutions. Vifor Pharma Group strives to help patients around the world with severe and chronic diseases lead better, healthier lives. The company develops, manufactures and markets pharmaceutical products for precision patient care. Vifor Pharma Group holds a leading position in all its core business activities and consists of the following companies: Vifor Pharma and Vifor Fresenius Medical Care Renal Pharma (a joint company with Fresenius Medical Care). Vifor Pharma Group is headquartered in Switzerland, and listed on the Swiss Stock Exchange (SIX Swiss Exchange, VIFN, ISIN: CH0364749348).
For more information, please visit viforpharma.com.
About Kissei Pharmaceutical Co., Ltd.
Kissei Pharmaceutical Co., Ltd. is a Japanese pharmaceutical company with approximately 70 years of history. Based on its management philosophy, “contributing to society through high-quality, innovative pharmaceutical products” and “serving society through our employees”, Kissei is concentrating on providing innovative pharmaceuticals to patients worldwide as a strongly R&D-oriented corporation. Kissei is engaged in R&D and licensing activities in the field of nephrology/dialysis, urology, and unmet medical needs in other disease areas. Kissei has an established collaboration with VFMCRP for sucroferric oxyhydroxide which Kissei fully developed in Japan as P-TOL® (known as Velphoro® in Europe/US) for the treatment of hyperphosphatemia. Since the launch in 2015, the market share of P-TOL® has been steadily expanding in Japan. For more information about Kissei Pharmaceutical, please visit www.kissei.co.jp.
About ChemoCentryx Inc.
ChemoCentryx is a biopharmaceutical company developing new medications for inflammatory and autoimmune diseases and cancer. ChemoCentryx targets the chemokine and chemoattractant systems to discover, develop and commercialize orally-administered therapies. Besides ChemoCentryx’s lead drug candidate, avacopan, ChemoCentryx also has early stage drug candidates that target chemoattractant receptors in other inflammatory and autoimmune diseases and in cancer.
About ANCA-associated vasculitis
ANCA-associated vasculitis is a systemic disease in which over-activation of the complement pathway further activates neutrophils, leading to inflammation and destruction of small blood vessels. This results in organ damage and failure, with the kidney as the major target, and is fatal if not treated. Currently, treatment for ANCA-associated vasculitis consists of courses of non-specific immuno-suppressants (cyclophosphamide or rituximab), combined with the administration of daily glucocorticoids (steroids) for prolonged periods of time, which can be associated with significant clinical risk including death from infection.
About TAVNEOS® (avacopan)
Avacopan is an orally-administered small molecule that is a selective inhibitor of the complement C5a receptor C5aR1. By precisely blocking the receptor (the C5aR) for the pro-inflammatory complement system fragment, C5a on destructive inflammatory cells such as blood neutrophils, avacopan arrests the ability of those cells to do damage in response to C5a activation, which is known to be the driver of inflammation. Moreover, avacopan’s selective inhibition of only the C5aR1 leaves the beneficial C5a l pathway through the C5L2 receptor functioning normally.
ChemoCentryx is also developing avacopan for the treatment of patients with C3 Glomerulopathy (C3G) and hidradenitis suppurativa (HS). The U.S. Food and Drug Administration has granted avacopan orphan-drug designation for ANCA-associated vasculitis, C3G and atypical hemolytic uremic syndrome. The European Commission has granted orphan medicinal product designation for avacopan for the treatment of two forms of ANCA vasculitis: microscopic polyangiitis and granulomatosis with polyangiitis (formerly known as Wegener’s granulomatosis), as well as for C3G. In October 2020, European Medicines Agency (EMA) accepted to review the Marketing Authorization Application (MAA) for avacopan for the treatment of patients with ANCA-associated vasculitis (granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA)).
On May 6, 2021 the U.S. Food & Drug Administration’s (FDA’s) Arthritis Advisory Committee narrowly voted in support of avacopan, a C5a receptor inhibitor, for the treatment of adult patients with anti-neutrophil cytoplasmic antibody (ANCA) associated vasculitis. Although the panelists were excited about the possibility of a steroid-sparing therapy, some raised questions about whether results from the single phase 3 trial could adequately inform the risk/benefit assessment.1 The FDA will weigh the panel’s recommendation as it considers possible approval.

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……………………………………………………………………………………………………………………………
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US323750645&_cid=P12-KU54VF-05767-1
Example 1: Preparation of Free Base Crystalline Form of Compound 1
Example 2: Preparing an Amorphous Form of Compound 1
PATENT
WO 2021163329
https://patents.google.com/patent/WO2021163329A1/en
PATENT
https://patents.google.com/patent/US20170283446A1/en
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US323750646&_cid=P12-KU54QB-03022-1
Example 1: A Besylate Salt of Compound 1 (Form I)
PATENT
WO 2011163640
https://patents.google.com/patent/WO2011163640A1

- Example 1
- [0097]
This example illustrates the preparation of (2R,3S)-2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methyl-benzoyl)-N-[4-methyl-3-(trifluoromethyl)phenyl]piperidine-3-carboxamide by the method provided more generally in FIG. 1 (Scheme 1) using the reagents provided below:
-

- [0098]
Step 1:
- [0099]
An oven-dried 12 L, 3-necked flask equipped with a mechanical stirrer, condenser, and thermometer was charged with acrolein diethyl acetal (1127 g, 8.666 mole, 1.05 equiv.) and warmed up to 40° C. A mixture of solid ethyl 3-(4-nitrophenyl)-3-oxo-propanoate (1956 g, 8.253 mole) and (R)-(−)-2-phenylglycinol (>99.5% e.e., 1187 g, 8.666 mole, 1.05 equiv.) was added in portions over 40 min. to maintain a stirrable mixture at an internal temperature of approximately 40° C. After all solids were added, the mixture was stirred at 40° C. for 10 minutes. 4M HCl in dioxane (206.2 mL, 0.825 mole, 10 mol. %) was subsequently added through the condenser within 2 minutes and the internal temperature was increased to 70 OC. The reaction was stirred for 22 h whereupon LC-MS showed consumption of starting materials and enamine intermediate. The heating was turned off and ethanol (6.6 L) was added. The solution was then seeded with 4 g of ethyl (3R,8aR)-5-(4-nitrophenyl)-3-phenyl-3,7,8,8a-tetrahydro-2H-oxazolo[3,2-a]pyridine-6-carboxylate and stirred at room temperature for 18 h. The solid was subsequently filtered off and 0.1 L of ethanol was used to rinse the flask and equipment onto the filter. The isolated solid was then washed three times on the filter with ethanol (250 mL each) and dried under vacuum to generate 1253 g of ethyl (3R,8aR)-5-(4-nitrophenyl)-3-phenyl-3,7,8,8a-tetrahydro-2H-oxazolo[3,2-a]pyridine-6-carboxylate as a bright yellow solid (38% yield, 98.5% HPLC wt/wt purity, 0.15 wt % of EtOH).
- [0100]
Step 2:
- [0101]
260 g of ethyl (3R,8aR)-5-(4-nitrophenyl)-3-phenyl-3,7,8,8a-tetrahydro-2H-oxazolo[3,2-a]pyridine-6-carboxylate (0.659 mol), 0.66 L of ethanol, and 56 g of palladium catalyst (10% Pd/C, Degussa type E101 NE/W, 50% wet, 21.5 wt. % of powder, 4.0 mol % Pd) were placed in a 2.2 L Parr bottle and purged with nitrogen. The bottle was mounted on a Parr shaker apparatus and hydrogen was added at a rate to keep the external temperature of the bottle below 30° C. After 4 hours, the consumption of hydrogen slowed down. The bottle was then shaken under 50 psi of hydrogen for 2 hours. 94 mL of glacial acetic acid (1.65 mol, 2.5 equiv.) was subsequently added to the bottle and the bottle was purged three times with hydrogen at 50 psi. The bottle was then shaken under 35-55 psi of hydrogen for 48 hours, keeping the temperature below 30° C. The bottle was removed from the apparatus and 55 mL of 12M HCl aq. was added (0.659 mol, 1 equiv.) followed by 87 mL of cyclopentanone (0.989 mol, 1.5 equiv.). The bottle was purged three times with hydrogen at 50 psi and then shaken under 50 psi of hydrogen for 16-20 hours. The mixture was removed from the apparatus and filtered through a fritted funnel containing celite (80 g) and then washed three times with 0.125 L of ethanol. 54.1 g of anhydrous sodium acetate (0.659 mol, 1 equiv.) was added and the mixture was concentrated in vacuo at 40-55° C. to remove 0.9 L of the volatile components. 2.0 L of acetonitrile was added and 2.0 L of volatile components were removed in vacuo. The crude material was diluted with 1.0 L of acetonitrile and mechanically stirred at r.t. for 30 minutes. The mixture was filtered through Celite (40 g) and the cake was washed with 0.28 L of acetonitrile. The combined filtrates gave a solution of the crude amine acetate (Solution A, e.e. =78%). Solutions A of two independent runs were combined for further processing.
- [0102]
In a 12-L 3-neck flask equipped with a mechanical stirrer, internal thermometer, and reflux condenser (−)-O,O′-di-p-toluoyl-L-tartaric acid (1.019 kg, 2.64 mol, 2 equiv.) was dissolved in 5.8 L of acetonitrile. The mixture was heated to 60° C. with stirring, followed by a quick addition of 1 L of Solution A. The resultant solution was seeded with 4 g of the crystalline ethyl (2R,3S)-2-[4-(cyclopentylamino)phenyl]piperidine-3-carboxylate (−)-O,O′-di-p-toluoyl-L-tartaric acid salt (1:2) and stirred at 60° C. for 15 minutes. After 15 minutes at 60 OC the seed bed has formed. The remaining amount of Solution A was added over a period of 2.5 hours, maintaining an internal temperature at 60° C. When the addition was complete, the heat source was turned off and the mixture was stirred for 17 hours, reaching a final temperature of 22.5° C. The suspension was filtered and the solids were washed with 0.50 L of acetonitrile to rinse the equipment and transfer all solids onto the filter. The resultant wet solids were washed on the funnel with 3.0 L of acetonitrile and dried in a vacuum oven at 45° C. for 48 hours to provide 1.005 kg of ethyl (2R,3S)-2-[4-(cyclopentylamino)phenyl]piperidine-3-carboxylate (−)-O,O′-di-p-toluoyl-L-tartaric acid salt (1:2) as an off-white solid (70% yield, contains 1 wt. % of acetonitrile). The enantiomeric ratio of the product was 99.4:0.6.
- [0103]
Step 3:
- [0104]
In a 5 L 3-necked flask equipped with a mechanical stirrer and an addition funnel, solid anhydrous potassium carbonate (K2CO3, 226 g, 1.64 mol, 4.1 equiv.) was dissolved in H2O (0.82 L) and cooled to ambient temperature. MTBE (0.82 L) was added, followed by solid ethyl (2R,3S)-2-[4-(cyclopentylamino)phenyl]piperidine-3-carboxylate (−)-O,O′-di-p-toluoyl-L-tartaric acid salt (1:2) (436 g, 0.400 mol). The mixture was vigorously stirred at r.t. for 1 hour, then 2-fluoro-6-methylbenzoyl chloride (72.5 g, 0.420 mmol, 1.05 equiv.) in MTBE (0.14 L) was added dropwise over 1 hour. The product started precipitating from the reaction before addition of the acid chloride was completed. The reaction was vigorously stirred at r.t. for 30 minutes and monitored by LC-MS for the disappearance of starting material. The mixture was subsequently transferred to a 5 L evaporation flask using 0.3 L of MTBE to rinse the equipment and remove all solids. The mixture was concentrated in vacuo to remove the MTBE, then 0.3 L of heptane was added and the mixture was evaporated again to leave only the product suspended in aqueous solution. The flask was removed from the rotavap and water (0.82 L) and heptane (0.82 L) were added. The suspension was vigorously stirred for 16 hours using a mechanical stirrer. The contents were then filtered and the solid was washed with water (2×0.42 L) and heptane (0.42 L). The solid was dried in a vacuum oven at 45° C. to provide 172 g of ethyl (2R,3S)-2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methyl-benzoyl)piperidine-3-carboxylate as an off-white powder (95% yield).
- [0105]
Step 4:
- [0106]
A 0.5 L 3-necked round-bottom flask was dried overnight in an oven at 200° C. and then cooled under a stream of nitrogen. The flask was equipped with a magnetic stir bar, nitrogen inlet, and a thermometer. The flask was charged with 30.2 g of ethyl (2R,3S)-2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methyl-benzoyl)piperidine-3-carboxylate (66.7 mmol), 11.5 mL of 4-methyl-5-trifluoromethylaniline (80 mmol, 1.2 equiv.) and 141 mL of dry toluene under an atmosphere of nitrogen. Nitrogen was bubbled through the resultant solution for 10 minutes and then the solution was warmed to 30° C. The oil bath was removed and 100 mL of a 2 M solution of AlMe3 in toluene (Aldrich, 200 mmol, 3 equiv.) was cannulated into the reaction mixture at a rate maintaining the reaction temperature between 35-40° C., a process that took approximately 45 minutes. The temperature of the reaction mixture was then increased to 55° C. over a period of 1 hour and the reaction mixture was stirred at 55° C. for 8 hours, whereupon all of the starting ester was consumed (monitored by LC-MS). The reaction was subsequently cooled overnight to ambient temperature and the solution was then cannulated into a mechanically stirred 1 L flask containing a solution of 67.8 g of sodium potassium tartrate tetrahydrate (240 mmol, 3.6 equiv.) in 237 mL of water, pre-cooled to 10 OC in an ice bath. The addition process took approximately 30 minutes, during which the reaction mixture self-heated to 57° C. The empty reaction flask was subsequently rinsed with 20 mL of dry toluene and the solution was combined with the quench mixture. The mixture was then cooled to r.t. with stirring, 91 mL of ethyl acetate was added, and the mixture was stirred an additional 15 minutes. The mixture was subsequently filtered through a pad of Celite and the filtrate was allowed to separate into two layers. The organic layer was then separated and washed with a solution of 5.7 g of sodium potassium tartrate tetrahydrate (20 mmol) in 120 mL of water and then with two 120 mL portions of water. The wet organic solution was concentrated in vacuo to a weight of ˜150 g and a solvent exchange with ethanol was performed maintaining a total volume of 0.2-0.3 L, until <1 mol. % toluene with respect to ethanol was observed by 1H NMR. The solution was then evaporated at elevated temperature to a weight of 223 g and heated to reflux. Mechanical stirring was initiated and 41 mL of water was added. The resulting solution was seeded with (2R,3S)-2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methyl-benzoyl)-N-[4-methyl-3-(trifluoromethyl)phenyl]piperidine-3-carboxamide crystals at 60 OC and then slowly cooled to r.t. over 2 hours. The slurry was subsequently stirred for 18 hours and the solids were filtered off. The solids were then washed with two 30 mL portions of 7:3 ethanol/water and dried in a vacuum oven for 24 hours at 50 OC to afford 31.0 g of (2R,3S)-2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methyl-benzoyl)-N-[4-methyl-3-(trifluoromethyl)phenyl]piperidine-3-carboxamide as off-white crystals (80% yield). Analytical data: HPLC purity: 99.59%; >99.8% d.e. and e.e. by HPLC; ICP-OES Pd: <1 ppm; Al: δ ppm; residual toluene by headspace GC-MS: 15 ppm; microash<0.1%; K—F 0.1%. 1H NMR (400 MHz, TFA-d) δ 7.91 (d, J=8.6 Hz, 1H), 7.84 (d, J=8.6 Hz, 1H), 7.58-6.82 (m, 8H), 6.75 (t, J=8.6 Hz, 1H), 4.10-4.00 (m, 1H), 3.60-3.47 (m, 1H), 3.45-3.41 (m, 1H), 3.33-3.25 (m, 1H), 2.44-2.22 (m, 7H), 2.04-1.92 (m, 4H), 1.82-1.69 (m, 7H), MS: (ES) m/z 582 (M+H+).
PATENT
WO2019236820
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019236820&_cid=P12-KU5444-91882-1
The present disclosure is directed to, inter alia, methods of treating ANCA-associated vasculitis (AAV) in a human in need thereof, the method comprising administering to the human a therapeutically effective amount of avacopan, having the structure shown below:
References
- Jayne DRW, Merkel PA, Schall TJ, et al. Avacopan for the treatment of ANCA-associated vasculitis. N Engl J Med. 2021 Feb 18;384(7):599–609.
- Merkel PA, Niles J, Jimenez R, et al. Adjunctive treatment with avacopan, an oral C5a receptor inhibitor, in patients with antineutrophil cytoplasmic antibody-associated vasculitis. ACR Open Rheumatol. 2020;2(11):662–671.
- Jayne DRW, Bruchfeld AN, Harper L, et al. Randomized trial of C5a receptor inhibitor avacopan in ANCA-associated vasculitis. J Am Soc Nephrol. 2017 Sep;28(9):2756–2767.
- U.S. Department of Health and Human Services. Food and Drug Administration. Demonstrative substantial evidence of effectiveness for human drug and biological products: Guidance for industry. 2019.
- Stone JH, Tuckwell K, Dimonaco S, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med. 2017 Jul 27;377(4):317–328.
- Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010 Jul 15;363(3):221–232.
- Warrington KJ. Avacopan—time to replace glucocorticoids? N Engl J Med. 2021 Feb 18;384(7):664–665.
////////////Avacopan, アバコパン , JAPAN 2021, APPROVALS 2021, CCX 168, авакопан , أفاكوبان , 阿伐可泮 ,
CC1=C(C(=CC=C1)F)C(=O)N2CCCC(C2C3=CC=C(C=C3)NC4CCCC4)C(=O)NC5=CC(=C(C=C5)C)C(F)(F)F