
















AZD 8931, Sapitinib, SAPATINIB
PHASE 2, at AstraZeneca for the treatment of non-small cell lung cancer.
CAS 848942-61-0,
MF C23H25ClFN5O3, MW 473.9,
pan-EGFR/pan-erbB inhibitor
4-[[4-[(3-Chloro-2-fluorophenyl)amino]-7-methoxy-6-quinazolinyl]oxy]-N-methyl-1-piperidineacetamide
4-(3-Chloro-2-fluoroanilino)-7-methoxy-6-[[1-(N-methylcarbamoylmethyl)piperidin-4-yl] oxy]quinazoline
4-(3-Chloro-2-fluoroanilino)-7-methoxy-6-[[1-(N-methylcarbamoylmethyl)piperidin-4-yl]oxy]quinazoline
2-[4-[4-(3-Chloro-2-fluoro-anilino)-7-methoxy-quinazolin-6-yl]oxy-1-piperidyl]-N-methyl-acetamide
AZD8931 is an oral, equipotent inhibitor of ErbB1, ErbB2 and ErbB3 receptor signaling.
WO 2005028469
| Inventors | Robert Hugh Bradbury, Laurent Francois Andre Hennequin, Bernard Christophe Barlaam, |
| Applicant | Astrazeneca Ab, Astrazeneca Uk Limited |
 Deregulation of the HER receptor family, comprising four related receptor tyrosine kinases (EGFR, HER2, HER3, and HER4), promotes proliferation, invasion, and tumor cell survival.Such deregulation has been observed in many human cancers, including lung, head and neck, and breast. Numerous small molecules have been investigated for inhibition of tyrosine kinases with the aminoquinazoline motif coming to the forefront as a privileged scaffold. Three of the clinically available treatments, gefitinib (1),lapatinib (2), and erlotinib (3),as well as the candidate drug dacomitinib (4), contain this arrangement
Deregulation of the HER receptor family, comprising four related receptor tyrosine kinases (EGFR, HER2, HER3, and HER4), promotes proliferation, invasion, and tumor cell survival.Such deregulation has been observed in many human cancers, including lung, head and neck, and breast. Numerous small molecules have been investigated for inhibition of tyrosine kinases with the aminoquinazoline motif coming to the forefront as a privileged scaffold. Three of the clinically available treatments, gefitinib (1),lapatinib (2), and erlotinib (3),as well as the candidate drug dacomitinib (4), contain this arrangement

Figure 1. Structure of gefitinib (1), lapatinib (2), erlotinib (3), dacomitinib (4), and AZD8931 (5).
SYNTHESIS
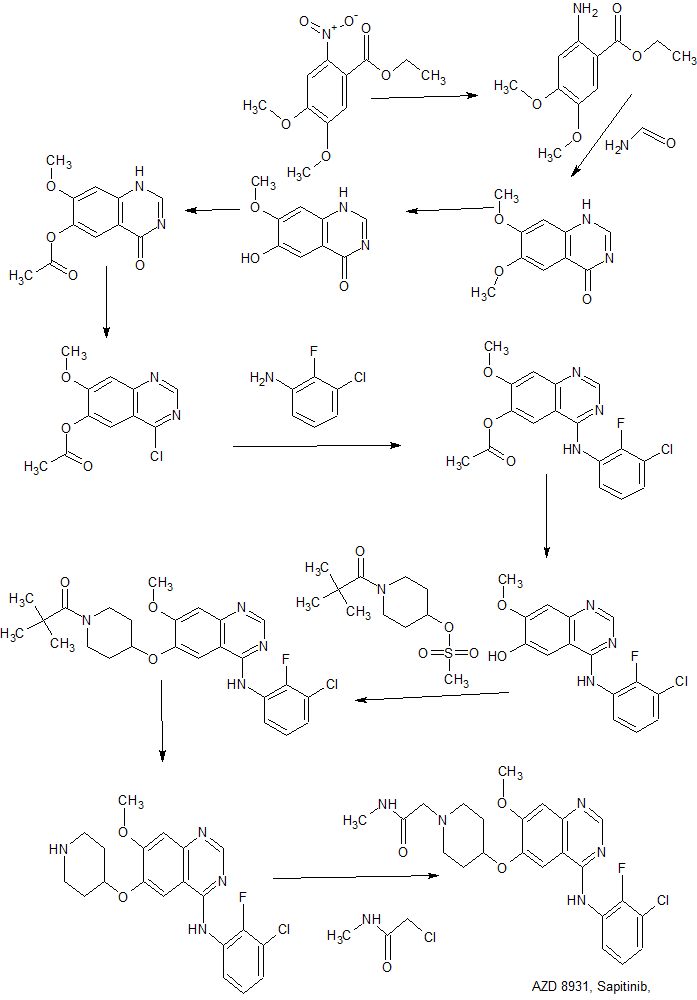
PATENT
https://www.google.com/patents/WO2005028469A1?cl=en
PAPER
The first radiosynthesis of [11C]AZD8931 as a new potential PET agent for imaging of EGFR, HER2 and HER3 signaling
Bioorganic & Medicinal Chemistry Letters (2014), 24, (18), 4455-4459.


Synthesis of the reference standard AZD8931 (11a)
Reagents and conditions: (a) SnCl2·H2O, concd HCl; (b) formamide, 168–170 °C; (c) l-methionine, methanesulfonic acid, 120 °C; (d) Ac2O, pyridine, DMAP, 100 °C; (e) POCl3, DEA, 100 °C; (f) 3-chloro-2-fluoroaniline, i-PrOH, refluxing; (g) conc. NH3, MeOH; (h) (1) Boc2O, CH2Cl2, dioxane; (2) methanesulfonyl chloride, Et3N, CH2Cl2; (i) Compound 8, CsF, DMA, 85 °C; (j) TFA; (k) Compound 11a: 2-chloro-N-methylacetamide, KI, K2CO3, CH3CN, refluxing; compound
PAPER
Discovery of AZD8931, an Equipotent, Reversible Inhibitor of Signaling by EGFR, HER2, and HER3 Receptors
ACS Medicinal Chemistry Letters (2013), 4, (8), 742-746.
Discovery of AZD8931, an Equipotent, Reversible Inhibitor of Signaling by EGFR, HER2, and HER3 Receptors

Deregulation of HER family signaling promotes proliferation and tumor cell survival and has been described in many human cancers. Simultaneous, equipotent inhibition of EGFR-, HER2-, and HER3-mediated signaling may be of clinical utility in cancer settings where the selective EGFR or HER2 therapeutic agents are ineffective or only modestly active. We describe the discovery of AZD8931 (2), an equipotent, reversible inhibitor of EGFR-, HER2-, and HER3-mediated signaling and the structure–activity relationships within this series. Docking studies based on a model of the HER2 kinase domain helped rationalize the increased HER2 activity seen with the methyl acetamide side chain present in AZD8931. AZD8931 exhibited good pharmacokinetics in preclinical species and showed superior activity in the LoVo tumor growth efficacy model compared to close analogues. AZD8931 is currently being evaluated in human clinical trials for the treatment of cancer.
4-(3-Chloro-2-fluoroanilino)-7-methoxy-6-{[1-(N-methylcarbamoylmethyl)piperidin-4-yl]oxy}quinazoline
(2). 2 as a white solid (60%).1H NMR (CDCl3):
δ 1.98 (m, 2H), 2.08 (m, 2H), 2.46 (m, 2H), 2.85 (m, 2H), 2.87 (d, 3H), 3.07 (s, 2H), 4.02 (s, 3H), 4.49 (m, 1H),
7.16 (m, 4H), 7.31 (m, 2H), 8.49 (m, 1H), 8.71 (s, 1H). MS-ESI m/z MH+ 474 [MH]+. Anal.
(C23H25ClFN5O3
.0.21 H2O) C, H, N. Found C, 57.88; H, 5.45; N, 14.67; Requires C, 57.83; H, 5.36; N, 14.66%.
PATENT
Compound (I) is disclosed in International Patent Application Publication number WO2005/028469 as Example 1 therein and is of the structure:

Compound (I)
Compound (I) is an erbB receptor tyrosine kinase inhibitor, in particular compound (I) is a potent inhibitor of EGFR and erbB2 receptor tyrosine kinases. Compound (I) also inhibits erbB3 mediated signalling through the inhibition of phosphorylation of erbB3 following ligand stimulated EGFR/erbB3 and/or erbB2/erbB3 heterodimerisation. Compound (I) is expected to be useful in the treatment of hyperproliferative disorders such as cancer.
WO 03/082831 discloses the preparation of various 4-(3-chloro-2- fluoroanilino)quinazo lines. However, compound (I) is not disclosed therein.
WO2005/028469 discloses as Example 1 therein the preparation of compound (I) as follows: 2-Chloro-N-methylacetamide (32 mg, 0.3 mmol) was added to a mixture of
4-(3-chloro-2-fluoroanilino)-7-methoxy-6-[(piperidin-4-yl)oxy]quinazoline (120 mg, 0.3 mmol), potassium iodide (16 mg, 0.1 mmol), and potassium carbonate (50 mg, 0.36 mmol) in acetonitrile (5 ml). The mixture was heated at reflux for one hour. After evaporation of the solvents under vacuum, the residue was taken up in dichloromethane. The organic solution was washed with water and brine, dried over magnesium sulfate. After evaporation of the solvents under vacuum, the residue was purified by chromatography on silica gel (eluant: 1% to 2% 7 N methanolic ammonia in dichloromethane) to give compound (I).
Scheme 1 :

Example 1 : Preparation of 4-(3-Chloro-2-fluoroanilino)-7-methoxy-6-{[l-(N- methylcarbamoylmethyl)piperidin-4-yl]oxy } quinazoline (Compound (I)).
Compound (I) was prepared according to the scheme shown below:

Compound (III) Compound (IV)
Compound (V)

Compound (I)Compound (II)
Step 1. Preparation of tert-butyl 4-(5-cyano-2-methoxyphenoxy)piperidine-l- carboxylate (Intermediate 2). 3-hydroxy-4-methoxybenzonitrile (Compound (X), 6.00 g, 39.62 mmole), tert-butyl (4-methanesulfonyloxy)piperidine-l-carboxylate (16.6 g, 59.44 mmoles) (Chemical & Pharmaceutical Bulletin 2001, 49(7), 822-829); and potassium carbonate (6.71 g, 47.55 mmoles) were suspended in isopropanol (78.98 g) and the mixture was heated at reflux with stirring. Additional tert-butyl (4-methanesulfonyloxy)piperidine-l- carboxylate (2.08 g, 7.43 mmoles) was added to push the reaction to completion. The mixture was then cooled and quenched by the addition of water (100.47 g). Seeding with intermediate 2 followed by cooling to 00C resulted in a crystalline product, which was isolated by filtration. The filter cake was washed with a mixture of water (8.86 g) and isopropanol (6.97 g), followed by water (23.64 g) and then dried to give Intermediate 2 (10.75 g, 80% yield); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.39 (s, 9 H) 1.48 (m, 2 H) 1.88 (m, 2 H) 3.13 (m, 2 H) 3.67 (m, 2 H) 3.83 (s, 3 H) 4.56 (tt, J=8.1, 3.8 Hz, 1 H) 7.13 (d, J=8.4 Hz, 1 H) 7.42 (dd, J=8.4, 1.9 Hz, 1 H) 7.51 (d, J=1.9 Hz, 1 H); Mass Spectrum: m/z (M + H)+ 333.1. Step 2. Preparation of 4-methoxy-3-(piperidin-4-yloxy)benzonitrile (Compound
(VI)). Intermediate 2 (39.31 g, 118.26 mmoles) was suspended in ethanol (155.53 g) and heated to 40 0C. To this slurry was slowly added HCl (46.61 g, 573.04 mmoles). The mixture was heated to 60 0C and held for 3 hours. The reaction mixture was cooled to 200C and seed was charged initiating crystallisation. The resulting solid was isolated by filtration at 00C, washed twice with ethanol (62.21 g) and then dried to give compound (VI) as the hydrochloride salt (29.84 g, 77% yield); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.84 (m, 2 H) 2.09 (m, 2 H) 3.02 (ddd, J=12.7, 8.9, 3.4 Hz, 2 H) 3.20 (m, 2 H) 3.84 (s, 3 H) 4.63 (tt, J=7.7, 3.6 Hz, 1 H) 7.15 (d, J=8.5 Hz, 1 H) 7.45 (dd, J=8.5, 1.9 Hz, 1 H) 7.56 (d, J=1.9 Hz, 1 H) 9.16 (br. s, 2 H); Mass Spectrum: m/z (M + H)+ 233.2. Step 3. Preparation of 2-[4-(5-cyano-2-methoxyphenoxy)piperidin-l-yl]-JV- methylacetamide (Compound (V)). Compound (VI) (28.36 g, 95.82 mmoles), 2-chloro-N- methylacetamide (12.37 g, 114.98 mmoles) and potassium carbonate (33.11 g, 239.55 mmoles) were suspended in acetonitrile (161.36 g). The reaction mixture was heated at reflux for 3 hours. The reaction mixture was cooled to 200C and water (386.26 g) was charged. The reaction was heated to 75°C and the volume reduced by distillation. Upon cooling crystallisation occurred. The resulting solid was isolated by filtration, washed twice with water (77.25 g and 128.75 g) and then dried to give compound (V) (27.95 g, 94% yield); 1H NMR (400 MHz, DMSO-J6) δ ppm 1.68 (m, 2 H) 1.91 (m, 2 H) 2.29 (m, 2 H) 2.61 (d, J=4.7 Hz, 3 H) 2.67 (m, 2 H) 2.88 (s, 2 H) 3.83 (s, 3 H) 4.41 (tt, J=8.3, 4.0 Hz, 1 H) 7.11 (d, J=8.4 Hz, 1 H) 7.40 (dd, J=8.4, 1.9 Hz, 1 H) 7.47 (d, J=I.9 Hz, 1 H) 7.68 (q, J=4.7 Hz, 1 H); Mass Spectrum: m/z (M + H)+ 304.2.
Step 4. Preparation of 2-[4-(5-cyano-2-methoxy-4-nitrophenoxy)piperidin-l-yl]-N- methylacetamide (Compound (IV)). Compound (V) (8.78 g, 26.11 mmoles) was suspended in acetic acid (22.82 g, 364.87 mmoles) and the resulting reaction mixture cooled to 5°C. To this was added sulfuric acid (23.64 g, 234.95 mmoles) maintaining the reaction temperature below 300C. To the resulting solution was added nitric acid (2.40 g, 26.63 mmoles). The reaction mixture was then heated to 35°C and held for 3 hours. Additional nitric acid (117 mg, 1.31 mmoles) and sulphuric acid (1.31 g 13.1 mmoles) were charged and the reaction mixture was heated at 35°C for 30 minutes. The solution was cooled to 200C and quenched with aqueous ammonia (92.45 g 1.36 moles), resulting in an increase in temperature to 500C. To the resulting slurry was added, propionitrile (61.58 g 1.12 moles) and water (19 g). The reaction mixture was heated to 80 0C resulting in a clear solution, which upon settling gave two layers. The bottom layer was removed. The reaction mixture was cooled to 20 0C resulting in a thick slurry. The solid was isolated by filtration, washed with propionitrile (6.16 g 112.0 mmoles) and dried to afford compound (IV) (7.44 g, 82% yield); 1H NMR (400 MHz, DMSO-de) δ ppm 1.72 (m, 2 H) 1.97 (m, 2 H) 2.35 (m, 2 H) 2.61 (d, J=4.7 Hz, 3 H) 2.66 (m, 2 H) 2.90 (s, 2 H) 3.96 (s, 3 H) 4.73 (tt, J=8.4, 4.0 Hz, 1 H) 7.71 (q, J=4.7 Hz, 1 H) 7.82 (s, 1 H) 7.86 (s, 1 H). Mass Spectrum: m/z (M + H)+ 349.2
Step 5. Preparation of 2-[4-(4-amino-5-cyano-2-methoxyphenoxy)piperidin-l-yl]-N- methylacetamide (Compound (III)). Compound (IV) (7.42 g, 19.38 mmoles) was suspended in water (44.52 g) and methanol (5.35 g). To this was added sodium dithionite (11.91 g, 58.15 mmoles) and the resulting reaction mixture was heated to 600C. To the reaction mixture was added hydrochloric acid (46.98 g, 463.89 mmoles)), resulting in a solution, which was held at 60 0C for 3 hours. The reaction mixture was then allowed to cool to 20 0C. Aqueous sodium hydroxide (15.51 g 182.2 mmoles) was charged followed by 2-methyltetrahydrofuran (58.0 g). The reaction mixture was heated to 60 0C, which upon settling gave two layers and the lower aqueous layer was discarded. The volume of the reaction mixture was reduced by vacuum distillation and methyl tert-butyl ether (18.54 g) was added to give a slurry which was cooled to 10 0C. and then the solid was collected by filtration. The solid was washed with 2- methyltetrahydrofuran (5.8 g) and dried to give compound (III) (5.4 g, 78% yield); 1H NMR (400 MHz, DMSO-de) δ ppm 1.62 (m, 2 H) 1.82 (m, 2 H) 2.20 (m, 2 H) 2.60 (d, J=4.7 Hz, 3 H) 2.65 (m, 2 H) 2.86 (s, 2 H) 3.72 (s, 3 H) 4.00 (tt, J=8.3, 4.0 Hz, 1 H) 5.66 (br. s, 2 H) 6.39 (s, 1 H) 6.94 (s, 1 H) 7.65 (q, J=4.7 Hz, 1 H). Mass Spectrum: m/z (M + H)+ 319.2.
Step 6. Preparation of 2-[4-(5-cyano-4-{[(dimethylamino)methylene]amino}-2- methoxyphenoxy)piperidin-l-yl]-Λ/-methylacetamide (Compound (H)). Compound (III) (18.21 g, 52.05 mmoles) was suspended in 2-methyltetrahydrofuran (99.62 g). To this was added acetic acid (162.79 mg), and N,N-dimethylformamide dimethyl acetal (DMA) (8.63 g, 70.27 mmoles) and the resulting reaction mixture was heated at 76 0C for 16 hrs. Additional N,N-dimethylformamide dimethyl acetal (639.41 mg, 5.20 mmoles) was added to the reaction mixture to ensure the reaction completed. The reaction mixture was cooled to 300C during which time crystallisation occurred. The resulting solid was isolated by filtration, washed with 2-methyltetrahydrofuran (14.23 g) and dried to afford compound (II) (19.53 g, 97% yield); 1H NMR (400 MHz, DMSO-J6) δ ppm 1.65 (m, 2 H) 1.86 (m, 2 H) 2.24 (m, 2 H) 2.60 (d, J=4.7 Hz, 3 H) 2.66 (m, 2 H) 2.87 (s, 2 H) 2.95 (s, 3 H) 3.04 (s, 3 H) 3.81 (s, 3 H) 4.19 (tt, J=8.2, 3.8 Hz, 1 H) 6.72 (s, 1 H) 7.15 (s, 1 H) 7.67 (q, J=4.7 Hz, 1 H) 7.90 (s, 1 H); Mass Spectrum: m/z (M + H)+ 374.2.
Step 7. Preparation of compound (I). 2-[4-(5-cyano-4-
{ [(dimethylamino)methylene] amino } -2-methoxyphenoxy)piperidin- 1 -yl] -JV-methylacetamide (compound (II), 7.00 g, 17.71 mmoles), was suspended in methoxybenzene (35.8 g). Acetic acid (16.6 g) was charged and to the resulting solution was added 3-chloro-2-fluoroaniline (2.71 g, 18.07 mmoles). The reaction mixture was heated at 90 0C for 20 hours then cooled to 200C. Water (37.04 g) was charged to the reaction mixture, and the organic layer discarded. To the resulting aqueous mixture was charged isopropanol (39.00 g), followed by aqueous ammonia (20.79 g, 25%). The reaction mixture was heated to 30 0C and seeded with compound (I), which induced crystallisation. The reaction was then cooled to 00C and the product isolated by filtration. The filter cake was washed twice with a mixture of water (7.28 g) and isopropanol (4.68 g), then dried to afford the compound (I) (5.65 g, 55% yield); 1H NMR (400 MHz, DMSO-J6) δ ppm 1.79 (m, 2 H) 2.04 (m, 2 H) 2.38 (m, 2 H) 2.62 (d, J=4.5 Hz, 3 H) 2.74 (m, 2 H) 2.94 (s, 2 H) 3.93 (s, 3 H) 4.56 (tt, J=8.1, 3.8 Hz, 1 H) 7.21 (s, 1 H) 7.28 (m, 1 H) 7.50 (m, 2 H) 7.73 (q, J=4.5 Hz, 1 H) 7.81 (s, 1 H) 8.36 (s, 1 H) 9.56 (br.s, 1 H); Mass Spectrum: m/z (M + H)+ 474.2, 476.2.
Example 2: Preparation of 4-(3-Chloro-2-fluoroanilino)-7-methoxy-6-{[l-(N- methylcarbamoylmethyl)piperidin-4-yl]oxy } quinazoline (Compound (I)). Compound (I) was prepared according to the scheme shown below:

Compound (III) Compound (IV)
Compound (V)

Compound (Xl)

Compound (I)
Steps 1, 2, 3 and 4 as set forth in Example 1.
Step 5, alternate 1. Preparation of compound (III). 2-[4-(5-Cyano-2-methoxy-4- nitrophenoxy)piperidin-l-yl]-N-methylacetamide (compound (IV), 15.00 g, 42.50 mmoles) was suspended in water (90.00 g) and methanol (59.38 g). To this was added sodium dithionite (30.47 g, 148.75 mmoles) and water (90.00 g), the resulting reaction mixture was heated to 30 0C and held for 2 hrs. To the reaction mixture was added hydrochloric acid (27.98 g, 276.25 mmoles)), resulting in a solution, which was held at 600C for 2 hours. Aqueous sodium hydroxide (30.60 g 382.49 mmoles) was added followed by a line wash of water (30.00 g). The reaction mixture was cooled to 25°C to give a slurry which was collected by filtration. The solid was washed with water (30.00 g) and dried to give compound (III) (13.50 g, 82% yield); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.62 (m, 2 H) 1.82 (m, 2 H) 2.20 (m, 2 H) 2.60 (d, J=4.7 Hz, 3 H) 2.65 (m, 2 H) 2.86 (s, 2 H) 3.72 (s, 3 H) 4.00 (tt, J=8.3, 4.0 Hz, 1 H) 5.66 (br. s, 2 H) 6.39 (s, 1 H) 6.94 (s, 1 H) 7.65 (q, J=4.7 Hz, 1 H). Mass Spectrum: m/z (M+H)+ 319.2.
Step 5, alternate 2. Preparation of compound (III). Compound (IV) (8.00 g, 22.67 mmoles) and 1% platinum + 2 % vanadium catalyst on carbon (1.23 g, 0.023 mmoles) were suspended in Acetonitrile (94.00 g). The reaction mixture was hydrogenated at a pressure of 3 Bar G and at a temperature of 35°C for 3 hrs. Once complete, the reaction mixture was filtered to remove the catalyst which is washed with acetonitrile (31.33 g). The volume of the reaction mixture was reduced by vacuum distillation to give a slurry which was cooled to 00C and then the solid was collected by filtration. The solid was washed with acetonitrile (12.53 g) and dried to give compound (III) (5.88 g, 78% yield); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.62 (m, 2 H) 1.82 (m, 2 H) 2.20 (m, 2 H) 2.60 (d, J=4.7 Hz, 3 H) 2.65 (m, 2 H) 2.86 (s, 2 H) 3.72 (s, 3 H) 4.00 (tt, J=8.3, 4.0 Hz, 1 H) 5.66 (br. s, 2 H) 6.39 (s, 1 H) 6.94 (s, 1 H) 7.65 (q, J=4.7 Hz, 1 H). Mass Spectrum: m/z (M+H)+ 319.2.
Step 6. Preparation of N, ΛT-bis(3-chloro-2-fluorophenyl)imidoformamide (compound (XI)). 3-chloro-2-fluroaniline (51.21 g, 341.22 mmoles) was suspended in cyclohexane (87.07 g). To this ethyl orthoformate (22.28 g, 150.32 mmoles) and acetic acid (0.94 g, 15.03 mmoles) were added. The resulting reaction mixture was heated, with stirring, to 48°C for 12 hours. Following this the reaction mixture was cooled to 200C over 12 hours and the solid product was isolated by filtration. The filter cake was washed with cylcohexane (26.12 g) and dried in vacuo at 40 0C to give compound (XI) as a white crystalline product (33.95 g, 93% yield); IH NMR Spectrum (400 MHz, DMSO-d6) δ ppm 7.14 (t, 2 H) 7.22 (m, 2 H) 8.14 (s, 1 H), 9,98 (s, 1 H); Mass Spectrum (by GC-MS EI): m/z (M+) 300.0.
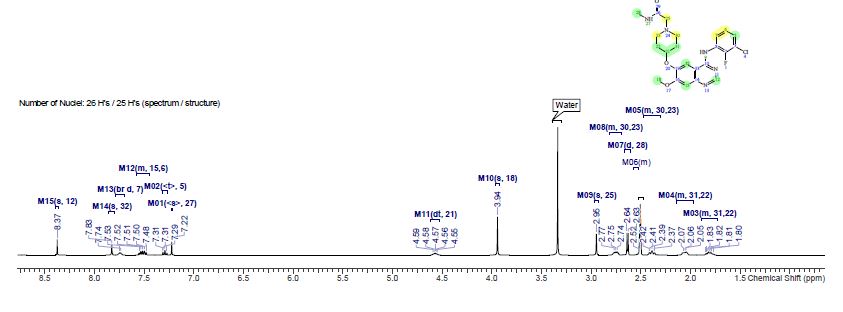
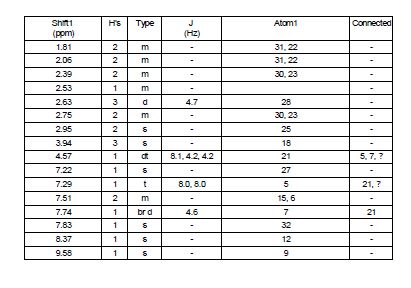
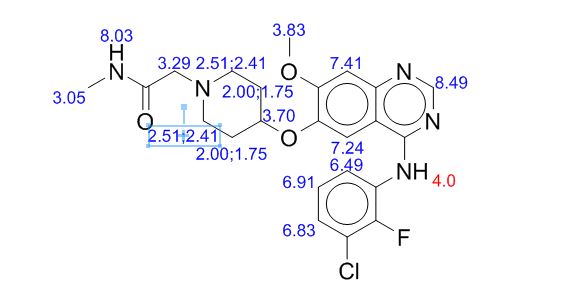
Step 7, alternate 1 : Preparation of compound (I). 2-[4-(4-Amino-5-cyano-2- methoxyphenoxy)piperidin-l-yl]-N-methylacetamide (compound (III)) (10 g, 29.84 mmol) and TV, ΛT-bis(3-chloro-2-fluorophenyl)imidoformamide (compound (XI)) (11.46 g, 37.3 mmol) were suspended in 2-methyltetrahydrofuran (30.4 ml) and heated to 800C. To this yellow suspension was added acetic acid (7.6 ml, 127.33 mmol) and the resulting solution was heated to 92°C for 6 hours. 2-methyltetrahydrofuran (66.5 ml) and water (28.5 ml) were added and mixture was cooled to 550C before adding 50%w/w sodium hydroxide (7 ml, 131.29 mmol) resulting in a temperature rise to 63°C. The temperature was raised further to 69°C and after settling the aqueous phase was discarded. The organic phase was washed with water (3 x 20 ml) and each aqueous phase was discarded after settling. 2- methyltetrahydrofuran (100 ml, 997 mmol) was added and the volume reduced by distillation. Seed was added to induce crystallisation and the resulting mixture was cooled to 15°C. The crystalline form was initially obtained following a spontaneous crystallisation from the experiment as described. The resulting solid was isolated by filtration, washed twice with 2- methyltetrahydrofuran (19 ml) and dried under vacuum at 400C to yield compound (I) as a white solid (12.14 g, 95%). 1H NMR (400 MHz, DMSO-J6) δ ppm 1.12 (d, J= 6Hz, 1.3H), 1.26 -1.36 (m, 0.4H), 1.75-1.97 (m, 3.3H), 2.02-2.15 (m, 2H), 2.35-2.44 (m, 2H), 2.64 (d, J= 4.7Hz, 3H), 2.72-2.80 (m, 2H), 2.95 (s, 2H), 3.52-3.59 (m, 0.4H), 3.72-3.87 (m, 0.86H), 3.95 (s, 3H), 4.53-4.63 (m, IH), 7.22 (s, IH), 7.29 (dt J= IHz J= 8Hz, IH), 7.51 (dt J= 7.4Hz, J= 18Hz, 2H), 7.71-7.77 (m, IH), 7.82 (s, IH), 8.37 (s, IH), 9.57 (s, IH). Mass Spectrum: m/z (M+H)+ 474.0. The NMR data above includes signals for the 2-methyltetrahydrofuran solvent which is present in a 0.43 molar equivalence. The signals pertaining to the solvent are at δ ppm shifts of 1.12, 1.26-1.36, 3.52-3.59 and 3.72-3.87. The cluster at 1.75-1.93 contains signals for the solvent and the parent compound. The XRPD for this compound is shown in Figure 2.
Step 7, alternate 2. Preparation of compound (I). Compound (III) (15 g, 44.76 mmol) and compound (XI) (17.19 g, 55.95 mmol) were suspended in 2-methyltetrahydrofuran (45.6 ml) and heated to 83°C. To this yellow suspension was added acetic acid (11.4 ml, 190.99 mmol) and the resulting solution was heated to 92°C for 3 Vi hours. 2-methyltetrahydrofuran (105 ml) and water (50 ml) were added and mixture was cooled to 49°C before adding 50%w/w sodium hydroxide (10.74 ml, 201.4 mmol), resulting in a temperature rise to 62°C. The temperature was maintained at 62°C and after settling the aqueous phase was discarded. The organic phase was washed with water (3 x 30 ml) and each aqueous phase was discarded after settling. The mixture was cooled to 15°C and seed was added to induce crystallisation. The crystalline form was initially obtained following a spontaneous crystallisation from the experiment as described. The resulting solid was isolated by filtration, washed twice with 2- methyltetrahydrofuran (21 ml) and dried under vacuum at 400C to yield compound (I) as a white solid (20.12 g, 95%). 1H NMR (400 MHz, DMSO-J6) δ ppm 1.75-1.86 (m, 2H), 2.02- 2.15 (m, 2H), 2.35-2.44 (m, 2H), 2.64 (d, J= 4.7Hz, 3H), 2.72-2.80 (m, 2H), 2.95 (s, 2H), 3.95 (s, 3H), 4.53-4.63 (m, IH), 7.22 (s, IH), 7.29 (dt J= IHz J= 8Hz, IH), 7.51 (dt J= 7.4Hz, J= 18Hz, 2H), 7.71-7.77 (m, IH), 7.82 (s, IH), 8.37 (s, IH), 9.57 (s, IH). Mass Spectrum: m/z (M+H)+ 474.0. The XRPD for this compound is shown in Figure 3.
Step 7, alternate 3. Preparation of compound (I). Compound (III) (15.1 g, 45.06 mmol) and compound (XI) (17.31 g, 56.32 mmol) were suspended in 2-methyltetrahydrofuran (46 ml) and heated to 800C. To this yellow suspension was added acetic acid (12 ml, 458 mmol) and the resulting solution was heated to 92° C for 7 hours. 2-methyltetrahydrofuran (100 ml) and water (43 ml) were added and mixture was cooled to 59°C before adding 50%w/w sodium hydroxide (11 ml, 207 mmol), resulting in a temperature rise to 71.5°C. The temperature was adjusted to 69°C and the aqueous phase was discarded after settling. The organic phase was washed with water (2 x 43 ml) and each aqueous phase was discarded after settling. 2-methyltetrahydrofuran (72 ml) was removed by distillation at atmospheric pressure and was replaced by addition of isopropyl alcohol (72 ml). A further 72 ml of solvent was removed by distillation at atmospheric pressure and replaced by isopropyl alcohol (72 ml). Seed was added to induce crystallisation and the resulting mixture was cooled to 15°C. The solid was isolated by filtration, washed twice with isopropylalcohol (32 ml) and dried under vacuum at 400C to yield compound (I) as a white solid (20.86 g, 87%). 1H NMR (400 MHz, DMSO-J6) δ ppm 1.04 (d, J= 6Hz, 6H),1.75-1.88 (m, 2H), 2.02-2.15 (m, 2H), 2.35-2.44 (m, 2H), 2.64 (d, J= 4.7Hz, 3H), 2.72-2.80 (m, 2H), 2.95 (s, 2H), 3.73-3.84 (m, IH), 3.95 (s, 3H), 4.34 (d, J = 4.2Hz, IH), 4.53-4.63 (m, IH), 7.22 (s, IH), 7.29 (dt J= IHz J= 8Hz, IH), 7.51 (dt J= 7Hz, J= 18Hz, 2H), 7.71-7.77 (m, IH), 7.82 (s, IH), 8.37 (s, IH), 9.57 (s, IH). Mass Spectrum: m/z (M+H)+ 474.0. The NMR data include signals for 1 mole equivalent isopropanol present. The XRPD for this compound is shown in Figure 4.
Example 3. Preparation of 4-(3-Chloro-2-fluoroanilino)-7-methoxy-6-{[l-(N- methylcarbamoylmethyl)piperidin-4-yl]oxy } quinazoline di- [(2E)-but-2-enedioate] (compound (I) difumarate salt). Compound (I) difumarate salt was prepared according to the scheme shown below:

Compound (III) Compound (IV) Compound (V)

Compound (Xl)
Difumarate Compound (I)
Steps 1, 2, 3, 4, 5 and 6 were performed as set forth in Example 2. Step 7. Preparation of compound (I) difumarate salt. Compound (III) (17.90 mmoles) and N, ΛT-bis(3-chloro-2-fluorophenyl)imidoformamide (compound (XI)) (7.04 g, 23.27 mmoles) were suspended in tert-butyl alcohol (88.95 g). To this suspension fumaric acid (10.39 g, 89.52 mmoles) was added and the mixture was heated to 800C, with stirring, for 2.5 hrs. Water (11.40 g, 632.80 mmoles) was charged and the reaction continued for a further 21.5 hrs. The reaction was cooled to 200C over 12 hours, during which time crystallisation occurred. The resulting solid was isolated by filtration and was washed with a mixture of water (1.00) and tert-butyl alcohol (7.80 g) followed by a wash with a mixture of water (0.50 g) and tert-butyl alcohol (7.30 g). The solid was dried in vacuo at 40 0C to give compound (I) difumarate salt (8.17 g, 61.40%) as a mustard yellow powder; 1H NMR (400 MHz, DMSO- dβ) δ ppm 1.83 (m, 2 H, broad) 2.07 (m, 2 H, broad) 2.64 (d, J=5.0 Hz, 3 H) 2.80 (m, 2 H, broad) 3.03 (s, 2 H) 3.94 (s, 3 H) 4.58 (m, 1 H) 6.63 (s, 4 H) 7.22 (s, 1 H) 7.29 (td, J=8.5, 1.0 Hz, 1 H) 7.51 (m, 2 H) 7.82 (m, 2 H) 8.37 (s, 1 H); Mass Spectrum: m/z (M+H)+ 474.0. Example 4. Preparation of 4-(3-Chloro-2-fluoroanilino)-7-methoxy-6-{[l-(N- methylcarbamoylmethyl)piperidin-4-yl]oxy}quinazoline (compound (I)).
Compound (I) was prepared according to the scheme shown below:

Compound (III) Compound (IV)
Compound (V)

Compound (XII)

Compound (I)
Steps 1, 2, 3, 4 and 5 were performed as set forth in Example 2.
Step 6. Preparation of N’-(3-chloro-2-fluoro-phenyl)-N,N-dimethyl-formamidine (compound (XII)). 3-chloro-2-fluroaniline (5.30 g, 35.29 mmoles) was dissolved in 2- methyltetrahydrofuran (52.94 g). To this N,N-dimethylformamide dimethyl acetal (6.07 g, 49.41 mmoles) and acetic acid (0.11 g, 1.76 mmoles) were added. The resulting reaction mixture was heated, with stirring, to 76 0C for 3 hours. Following this the solvent was removed in vacuo at 400C to give compound (XII) as a yellow oil (6.60 g, 93% yield); IH NMR Spectrum (400 MHz, DMSO-d6) δ ppm 2.74 (s, 0.29H), 2.89 (s, 0.31H), 2.94 (s, 2.75H), 3.03 (s, 2.66H), 3.34 (br s, 0.70H), 5.48 (s, 0.06H) 6.91-7.10 (m, 3H), 7.79 (s, 1 H), 7.96 (s, 1 H). The NMR data above includes signals for N,N-dimethylformamide dimethyl acetal which is present in a 0.06 molar equivalence. The signals pertaining to N5N- dimethylformamide dimethyl acetal are at δ ppm shifts of 3.75, and 6.90-6.95. The signal at δ ppm 3.35 is due to residual water. Mass Spectrum (by LCMS EI): m/z (M+H)+ 201.2. Step 7: Preparation of compound (I). 2-[4-(4-Amino-5-cyano-2- methoxyphenoxy)piperidin-l-yl]-N-methylacetamide (compound (III)) (0.50 g, 1.45 mmol) and N’-(3-chloro-2-fluoro-phenyl)-N,N-dimethyl-formamidine (compound (XII)) (0.32 g, 1.52 mmol) were suspended in methoxybenzene (3.1 ml). To this yellow suspension was added acetic acid (1.52 ml, 25.51 mmol) and the resulting solution was heated to 90 0C for 14 hours. The reaction mixture was cooled to 20 0C and water (2.58 mL) was added. The organic layer was removed and the aqueous layer washed with methoxybenzene (1.4 mL). Ethanol (2.45 mL) and ammonia (1.94 ml, 25.55 mmoles) were added to the aqueous layer. The solution was heated to 900C resulting in the loss of some ethanol by evaporation. The solution was cooled to 40 0C. Seed was added to induce crystallisation and the resulting mixture was cooled to 20 0C. The solid was isolated by filtration to yield compound (I) as a white solid (0.61 g, 73% yield). IH NMR (400 MHz, DMSO-d6) δ ppm 1.75-1.87 (m, 2H), 2.02-2.15 (m, 2H), 2.35-2.44 (m, 2H), 2.64 (d, J= 4.8Hz, 3H), 2.72-2.80 (m, 2H), 2.95 (s, 2H), 3.35 (s, 5.4H), 3.75 (s, 1.3H), 3.95 (s, 3H), 4.58 (hept., J=4.0Hz, IH), 6.90-6.95 (m, 1.3H), 7.23 (s, 1.8H), 7.26-7.34 (m, IH), 7.45-7.58 (m 2H), 7.72-7.78 (m, IH), 7.83 (s, IH), 8.38 (s, IH), 9.58 (s, IH). The NMR data above includes signals for the methoxybenzene solvent which is present in a 0.40 molar equivalence. The signals pertaining to the solvent are at δ ppm shifts of 3.75, and 6.90-6.95. The cluster at 7.26-7.34 contains signals for the solvent and the parent compound. The signal at δ ppm 3.35 is due to residual water. Mass Spectrum: m/z (M + H)+ 474.0, 476.0. Example 5. Preparation of compound (I) difumarate Form A – 2-[4-({4-[(3-Chloro-2- fluorophenyl)amino]-7-methoxyquinazolin-6-yl}oxy)piperidin-l-yl]-N-methylacetamide di- [(2E)-but-2-enedioate] Form A. A solution of fumaric acid (2.7 g, 23.22 mmol) in methanol (95 ml) was added to a mixture of 2-[4-({4-[(3-Chloro-2-fluorophenyl)amino]-7- methoxyquinazolin-6-yl}oxy)piperidin-l-yl]-N-methylacetamide (compound (I)) (5.62 g at 89% w/w, 10.55 mmol) in isopropanol (100 ml) maintaining the temperature >65°C. The mixture was heated at reflux for one hour before clarification. The reaction mixture was cooled to 300C over 90 minutes and held for 30 minutes to establish crystallisation. The reaction was cooled to 00C over 2 hours and held for 1 hour before isolation by filtration. The filter cake was washed twice with cold isopropanol (2 x 10 ml) and dried in vacuo at 500C to give the title compound as a white solid (5.84 g, 78%); 1H NMR Spectrum: (DMSO) 1.85 (m, IH), 2.08 (m, IH), 2.50 (m, IH), 2.66 (d, 3H), 2.83 (m, IH), 3.05 (s, 2H), 3.96 (s, 3H), 4.58 (m, IH), 6.64 (s, 4H), 7.23 (s, IH), 7.28 (m, IH), 7.46 (ddd, IH), 7.55 (m, IH), 7.70 (broad q, IH), 7.85 (s, IH), 8.38 (s, IH).
Example 6. Preparation of compound (I) difumarate Form A: 2-[4-({4-[(3-Chloro-2- fluorophenyl)amino]-7-methoxyquinazolin-6-yl}oxy)piperidin-l-yl]-N-methylacetamide di- [(2E)-but-2-enedioate] Form A. A solution of fumaric acid (1.4 kg, 12.1 mol) in methanol (26.6 kg) was added to a mixture of 2-[4-({4-[(3-chloro-2-fluorophenyl)amino]-7- methoxyquinazolin-6-yl}oxy)piperidin-l-yl]-N-methylacetamide (2.93 kg, 84.8% w/w, 5.24 mol) in isopropanol (39 kg) maintaining the temperature >65°C. A line wash of methanol (3.6 kg) was charged. The mixture was heated at reflux for one hour before clarification, followed by a line wash of methanol (7 kg). The reaction mixture was distilled at atmospheric pressure to remove 47 kg of distillates. Isopropanol (15.8 kg was added and the reaction mixture distilled to remove 15.6 kg of distillates. Crystallisation occurred during the distillation. Isopropanol (21 kg) was added and the reaction cooled to 00C over 8 hours and held for 1 hour before isolation by filtration. The filter cake was washed with cold 50:50 isopropanol:MeOH (4 kg) followed by cold isopropanol (4 kg) and dried in vacuo at 500C to give the title compound as a white solid (3.64 kg, 98%); 1H NMR Spectrum: (DMSO) 1.85 (m, IH), 2.08 (m, IH), 2.50 (m, IH), 2.66 (d, 3H), 2.83 (m, IH), 3.05 (s, 2H), 3.96 (s, 3H), 4.58 (m, IH), 6.64 (s, 4H), 7.23 (s, IH), 7.28 (m, IH), 7.46 (ddd, IH), 7.55 (m, IH), 7.70 (broad q, IH), 7.85 (s, IH), 8.38 (s, IH).
Example 7. Preparation of compound (I) difumarate Form A: 2-[4-({4-[(3-Chloro-2- fluorophenyl)amino]-7-methoxyquinazolin-6-yl}oxy)piperidin-l-yl]-N-methylacetamide di- [(2E)-but-2-enedioate] Form A. 2-[4-({4-[(3-Chloro-2-fluorophenyl)amino]-7- methoxyquinazolin-6-yl}oxy)piperidin-l-yl]-N-methylacetamide (compound (I)) (60.19 g at 88% w/w, 111.8 mmol) was dissolved in ethyl acetate (1550 ml). The solution was clarified by filtration and the filter washed with ethyl acetate (53 ml). The solution was cooled to 400C. A clarified solution of fumaric acid (26.60 g, 257.0 mmol) in isopropanol (408 ml) was then added over 1 hour. The filter used to clarify the fumaric acid solution was then washed with isopropanol (37 ml). After holding for 1 hour at 400C the reaction was cooled to 200C over 1 hour. The reaction mixture was held for 13.5 hours before isolating the product by filtration. The filter cake was washed twice with ethyl acetate (82 ml) : isopropanol (24 ml) and then dried in vacuo at 400C to give the title compound as a white solid (72.32 g, 90%); 1H NMR Spectrum: (DMSO) 1.85 (m, IH), 2.08 (m, IH), 2.50 (m, IH), 2.66 (d, 3H), 2.83 (m, IH), 3.05 (s, 2H), 3.96 (s, 3H), 4.58 (m, IH), 6.64 (s, 4H), 7.23 (s, IH), 7.28 (m, IH), 7.46 (ddd, IH), 7.55 (m, IH), 7.70 (broad q, IH), 7.85 (s, IH), 8.38 (s, IH). Example 8. Preparation of compound (I) difumarate Form A: 2-[4-({4-[(3-Chloro-2- fluorophenyl)amino]-7-methoxyquinazolin-6-yl}oxy)piperidin-l-yl]-N-methylacetamide di- [(2E)-but-2-enedioate] Form A. 2-[4-({4-[(3-Chloro-2-fluorophenyl)amino]-7- methoxyquinazolin-6-yl}oxy)piperidin-l-yl]-N-methylacetamide (compound (I)) (2.75 g at assumed 100% w/w, 5.80 mmol) was dissolved in ethyl acetate (94 ml) and isopropanol (14 ml). The solution was distilled such that 25.2 ml of distillates were collected. The solution was cooled to 400C. A clarified solution of fumaric acid (1.38 g, 11.90 mmol) in isopropanol (21 ml) was then added over 1 hour. Compound (I) difumarate Form A seed was added (3.7 mg, 5.3 μmol). The filter used to clarify the fumaric acid solution was then washed with isopropanol (2 ml). After holding for 1 hour at 400C the reaction was cooled to 200C over 2 hours. The reaction mixture was held for 15 hours before isolating the product by filtration. The filter cake was washed twice with ethyl acetate (4.3 ml): isopropanol (1.2 ml) and then dried in vacuo at 400C to give the title compound as a white solid (72.32 g, 90%); 1H NMR Spectrum: (DMSO) 1.85 (m, IH), 2.08 (m, IH), 2.50 (m, IH), 2.66 (d, 3H), 2.83 (m, IH), 3.05 (s, 2H), 3.96 (s, 3H), 4.58 (m, IH), 6.64 (s, 4H), 7.23 (s, IH), 7.28 (m, IH), 7.46 (ddd, IH), 7.55 (m, IH), 7.70 (broad q, IH), 7.85 (s, IH), 8.38 (s, IH).
Example 9. Preparation of compound (I) difumarate Form A: 2-[4-({4-[(3-Chloro-2- fluorophenyl)amino]-7-methoxyquinazolin-6-yl}oxy)piperidin-l-yl]-N-methylacetamide di- [(2E)-but-2-enedioate] Form A. 2-[4-({4-[(3-Chloro-2-fluorophenyl)amino]-7- methoxyquinazolin-6-yl}oxy)piperidin-l-yl]-N-methylacetamide (compound (I)) (1 g, 1.86 mmoles) and fumaric acid (0.44 g, 3.81 mmoles) were suspended in water (4.4 g) and heated to 85°C. The reaction mixture was cooled to 600C at l°C/minute and compound (I) Form A seed was added when the temperature was 77°C. The resulting solid was isolated by filtration, washed twice with acetone (0.7O g per wash) and dried in a vacuum oven at 400C to afford the title compound (0.89 g, 68% yield), IH NMR (400 MHz, DMSO-d6) d ppm 1.84 (m, 2 H) 2.08 (m, 2 H) 2.55 (m, 2 H) 2.63 (d, J=4.7 Hz, 3 H) 2.86 (m, 2 H) 3.12 (s, 2 H) 3.93 (s, 3 H) 4.59 (tt, J=7.8, 3.7 Hz, 1 H) 6.62 (s, 4 H) 7.21 (s, 1 H) 7.27 (td, J=8.1, 1.3 Hz, 1 H) 7.49 (m, 2 H) 7.86 (m, 2 H) 8.36 (s, 1 H) 9.63 (br. s., 1 H). Compound (I) difumarate Form A is a free flowing powder.
PAPER
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.6b00412
The Development of a Dimroth Rearrangement Route to AZD8931
 , Kay Boardman‡, Oliver Cunningham‡, Matthew Evans‡, Martin F. Jones‡§, Kirsty Millard‡, Raquel Rozada-Sanchez‡, Yvonne Sawyer†, Paul Siedlecki‡, and Brian Whitlock‡
, Kay Boardman‡, Oliver Cunningham‡, Matthew Evans‡, Martin F. Jones‡§, Kirsty Millard‡, Raquel Rozada-Sanchez‡, Yvonne Sawyer†, Paul Siedlecki‡, and Brian Whitlock‡
Recently, the aminoquinazoline motif has been highly prevalent in anticancer pharmaceutical compounds. Synthetic methods are required to make this structure on a multikilo scale and in high purity. The initial route to aminoquinazoline AZD8931 suffered from the formation of late-stage impurities. To avoid these impurities, a new high-yielding Dimroth rearrangement approach to the aminoquinazoline core of AZD8931 was developed. Assessment of route options on a gram scale demonstrated that the Dimroth rearrangement is a viable approach. The processes were then evolved for large-scale production with learning from a kilo campaign and two plant-scale manufactures. Identification of key process impurities offers an insight into the mechanisms of the Dimroth rearrangement as well as the hydrogenation of a key intermediate. The final processes were operated on a 30 kg scale delivering the target AZD8931 in 41% overall yield.
2-[4-[4-(3-chloro-2-fluoro-anilino)-7-methoxy-quinazolin-6-yl]oxy-1-piperidyl]-N-methyl-acetamide IPA solvate (5) as a white solid (38.1 kg, 84.2% yield); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.81 (m, 2 H), 2.06 (m, 2 H), 2.39 (m, 2 H), 2.63 (d, J = 4.7 Hz, 3 H), 2.75 (m, 2 H), 2.95 (s, 2 H), 3.94 (s, 3 H), 4.57 (Dt, J = 8.1, 4.2 Hz, 1 H), 7.22 (s, 1 H), 7.29 (t, J = 8.0 Hz, 1 H), 7.51 (m, 2 H), 7.74 (br d, J = 4.6 Hz, 1 H), 7.83 (s, 1 H), 8.37 (s, 1 H), 9.58 (br.s, 1 H); m/Z ES+ 474.2 [MH]+; HRMS found [MH]+ = 474.1706, C23H25ClFN5O3 requires [MH]+ = 474.1630; Assay (QNMR) 97.5 wt %/wt.
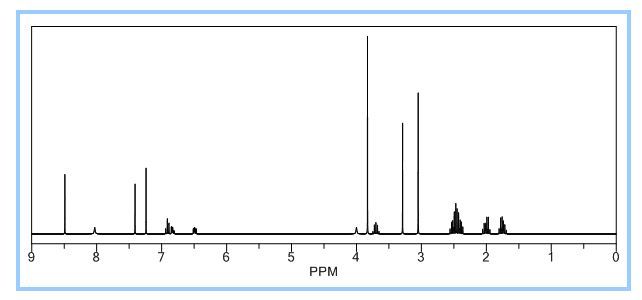
1H NMR PREDICT
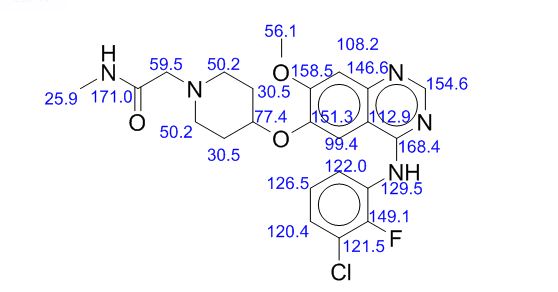
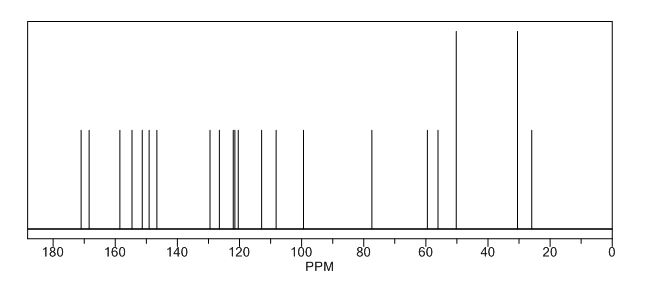
13C NMR PREDICT
| CHEMICAL & PHARMACEUTICAL BULLETIN, vol. 49, no. 7, 2001, pages 822 – 829 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2013051883A3 * | Oct 5, 2012 | Jun 6, 2013 | Hanmi Science Co., Ltd. | Method for preparing 1-(4-(4-(3,4-dichloro-2-fluorophenylamino)-7-methoxyquinazolin-6-yloxy)piperidin-1-yl)-prop-2-en-1-one hydrochloride and intermediates used therein |
| US8859767 | Oct 5, 2012 | Oct 14, 2014 | Hanmi Science Co., Ltd | Method for preparing 1-(4-(4-(3,4-dichloro-2-fluorophenylamino)-7-methoxyquinazolin-6-yloxy)piperidin-1-yl)-prop-2-en-1-one hydrochloride and intermediates used therein |
////////////////AZD 8931, Sapitinib, pan-EGFR, pan-erbB inhibitor, SAPATINIB, PHASE 2, 848942-61-0
CNC(=O)CN1CCC(CC1)OC2=C(C=C3C(=C2)C(=NC=N3)NC4=C(C(=CC=C4)Cl)F)OC
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This article is a compilation for educational purposes only.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent