














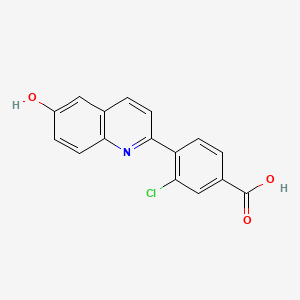
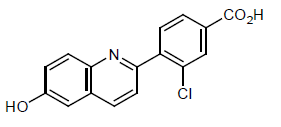
Cavosonstat (N-91115)
CAS 1371587-51-7
C16H10ClNO3, 299.71 g/mol
UNII-O2Z8Q22ZE4, O2Z8Q22ZE4, NCT02589236; N91115-2CF-05; SNO-6
3-chloro-4-(6-hydroxyquinolin-2-yl)benzoic acid
Treatment of Chronic Obstructive Pulmonary Diseases (COPD), AND Cystic fibrosis, Nivalis Therapeutics, phase 2
The product was originated at Nivalis Therapeutics, which was acquired by Alpine Immune Sciences in 2017. In 2018, Alpine announced the sale and transfer of global rights to Laurel Venture Capital for further product development.
In 2016, orphan drug and fast track designations were granted to the compound in the U.S. for the treatment of cystic fibrosis.
- Originator N30 Pharma
- Developer Nivalis Therapeutics
- Class Small molecules
- Mechanism of Action Cystic fibrosis transmembrane conductance regulator modulators; Glutathione-independent formaldehyde dehydrogenase inhibitors; Nitric oxide stimulants
- Orphan Drug Status Yes – Cystic fibrosis
- 20 Jul 2018 Laurel Venture Capital acquires global rights for cavosonstat from Alpine Immune Sciences
- 20 Jul 2018 Laurel Venture Capital plans a phase II trial for Asthma
- 24 Jun 2018 Biomarkers information updated
Cavosonstat, alos known as N91115) an orally bioavailable inhibitor of S-nitrosoglutathione reductase, promotes cystic fibrosis transmembrane conductance regulator (CFTR) maturation and plasma membrane stability, with a mechanism of action complementary to CFTR correctors and potentiators.
 Cavosonstat (N91115) was an experimental therapy being developed by Nivalis Therapeutics. Its primary mechanism of action was to inhibit the S-nitrosoglutathione reductase (GSNOR) enzyme and to stabilize cystic fibrosis transmembrane regulator (CFTR) protein activity. A press release published in February announced the end of research for this therapy in cystic fibrosis (CF) patients with F508del mutations. The drug, which did not meet primary endpoints in a Phase 2 trial, had been referred to as the first of a new class of compounds that stabilizes the CFTR activity.
Cavosonstat (N91115) was an experimental therapy being developed by Nivalis Therapeutics. Its primary mechanism of action was to inhibit the S-nitrosoglutathione reductase (GSNOR) enzyme and to stabilize cystic fibrosis transmembrane regulator (CFTR) protein activity. A press release published in February announced the end of research for this therapy in cystic fibrosis (CF) patients with F508del mutations. The drug, which did not meet primary endpoints in a Phase 2 trial, had been referred to as the first of a new class of compounds that stabilizes the CFTR activity.
History of cavosonstat
During preclinical studies, N91115 (later named cavosonstat) demonstrated an improvement in cystic fibrosis transmembrane regulator (CFTR) stability.
A Phase 1 study was initiated in 2014 to evaluate the safety, tolerability, and pharmacokinetics (how a drug is processed in the body) of the drug in healthy volunteers. Later that year, the pharmacokinetics of the drug were assessed in another Phase 1 trial involving CF patients with F508del mutation suffering from pancreatic insufficiency. Results were presented a year later by Nivalis, revealing good tolerance and safety in study participants.
A second, much smaller Phase 2 study (NCT02724527) assessed cavosonstat as an add-on therapy to ivacaftor (Kalydeco). This double-blind, randomized, placebo-controlled study included 19 participants who received treatment with cavosonstat (400 mg) added to Kalydeco or with placebo added to Kalydeco. The primary objective was change in lung function from the study’s start to week 8. However, the treatment did not demonstrate a benefit in lung function measures or in sweat chloride reduction at eight weeks (primary objective). As a result, Nivalis decided not to continue development of cavosonstat for CF treatment.
The U.S. Food and Drug Administration (FDA) had granted cavosonstat both fast track and orphan drug designations in 2016.
How cavosonstat works
The S-nitrosoglutathione (GSNO) is a signaling molecule that is present in high concentrations in the fluids of the lungs or muscle tissues, playing an important role in the dilatation of the airways. GSNO levels are regulated by the GSNO reductase (GSNOR) enzyme, altering CFTR activity in the membrane. In CF patients, GSNO levels are low, causing a loss of the airway function.
Cavosonstat’s mechanism of action is achieved through GSNOR inhibition, which was presumed to control the deficient CFTR protein. Preclinical studies showed that cavosonstat restored GSNO levels.
The chemical compound nitric oxide is a gas with chemical formula NO. NO is one of the few gaseous signaling molecules known in biological systems, and plays an important role in controlling various biological events. For example, the endothelium uses NO to signal surrounding smooth muscle in the walls of arterioles to relax, resulting in vasodilation and increased blood flow to hypoxic tissues. NO is also involved in regulating smooth muscle proliferation, platelet function, and neurotransmission, and plays a role in host defense. Although NO is highly reactive and has a lifetime of a few seconds, it can both diffuse freely across membranes and bind to many molecular targets. These attributes make NO an ideal signaling molecule capable of controlling biological events between adjacent cells and within cells.
[0003] NO is a free radical gas, which makes it reactive and unstable, thus NO is short lived in vivo, having a half life of 3-5 seconds under physiologic conditions. In the presence of oxygen, NO can combine with thiols to generate a biologically important class of stable NO adducts called S-nitrosothiols (SNO’s). This stable pool of NO has been postulated to act as a source of bioactive NO and as such appears to be critically important in health and disease, given the centrality of NO in cellular homeostasis (Stamler et al., Proc. Natl. Acad. Sci. USA, 89:7674-7677 (1992)). Protein SNO’s play broad roles in the function of cardiovascular, respiratory, metabolic, gastrointestinal, immune, and central nervous system (Foster et al., Trends in Molecular Medicine, 9 (4): 160-168, (2003)). One of the most studied SNO’s in biological systems is S-nitrosoglutathione (GSNO) (Gaston et al., Proc. Natl. Acad. Sci. USA 90: 10957-10961 (1993)), an emerging key regulator in NO signaling since it is an efficient trans-nitrosating agent and appears to maintain an equilibrium with other S-nitrosated proteins (Liu et al., Nature, 410:490-494 (2001)) within cells. Given this pivotal position in the NO-SNO continuum, GSNO provides a therapeutically promising target to consider when NO modulation is pharmacologically warranted.
[0004] In light of this understanding of GSNO as a key regulator of NO homeostasis and cellular SNO levels, studies have focused on examining endogenous production of GSNO and SNO proteins, which occurs downstream from the production of the NO radical by the nitric oxide synthetase (NOS) enzymes. More recently there has been an increasing understanding of enzymatic catabolism of GSNO which has an important role in governing available concentrations of GSNO and consequently available NO and SNO’s.
[0005] Central to this understanding of GSNO catabolism, researchers have recently identified a highly conserved S-nitrosoglutathione reductase (GSNOR) (Jensen et al., Biochem J., 331 :659-668 (1998); Liu et al., (2001)). GSNOR is also known as glutathione-dependent formaldehyde dehydrogenase (GSH-FDH), alcohol dehydrogenase 3 (ADH-3) (Uotila and Koivusalo, Coenzymes and Coƒactors., D. Dolphin, ed. pp. 517-551 (New York, John Wiley & Sons, (1989)), and alcohol dehydrogenase 5 (ADH-5). Importantly GSNOR shows greater activity toward GSNO than other substrates (Jensen et al., (1998); Liu et al., (2001)) and appears to mediate important protein and peptide denitrosating activity in bacteria, plants, and animals. GSNOR appears to be the major GSNO-metabolizing enzyme in eukaryotes (Liu et al., (2001)). Thus, GSNO can accumulate in biological compartments where GSNOR activity is low or absent (e.g. , airway lining fluid) (Gaston et al., (1993)).
[0006] Yeast deficient in GSNOR accumulate S-nitrosylated proteins which are not substrates of the enzyme, which is strongly suggestive that GSNO exists in equilibrium with SNO-proteins (Liu et al., (2001)). Precise enzymatic control over ambient levels of GSNO and thus SNO-proteins raises the possibility that GSNO/GSNOR may play roles across a host of physiological and pathological functions including protection against nitrosative stress wherein NO is produced in excess of physiologic needs. Indeed, GSNO specifically has been implicated in physiologic processes ranging from the drive to breathe (Lipton et al., Nature, 413: 171-174 (2001)) to regulation of the cystic fibrosis transmembrane regulator (Zaman et al., Biochem Biophys Res Commun, 284:65-70 (2001)), to regulation of vascular tone, thrombosis, and platelet function (de Belder et al., Cardiovasc Res.; 28(5):691-4 (1994)), Z. Kaposzta, et al., Circulation; 106(24): 3057 – 3062, (2002)) as well as host defense (de Jesus-Berrios et al., Curr. Biol., 13: 1963-1968 (2003)). Other studies have found that GSNOR protects yeast cells against nitrosative stress both in vitro (Liu et al., (2001)) and in vivo (de Jesus-Berrios et al., (2003)).
[0007] Collectively, data suggest GSNO as a primary physiological ligand for the enzyme S-nitrosoglutathione reductase (GSNOR), which catabolizes GSNO and
consequently reduces available SNO’s and NO in biological systems (Liu et al., (2001)), (Liu et al., Cell, 116(4), 617-628 (2004)), and (Que et al., Science, 308, (5728): 1618-1621 (2005)). As such, this enzyme plays a central role in regulating local and systemic bioactive NO. Since perturbations in NO bioavailability has been linked to the pathogenesis of numerous disease states, including hypertension, atherosclerosis, thrombosis, asthma, gastrointestinal disorders, inflammation, and cancer, agents that regulate GSNOR activity are candidate therapeutic agents for treating diseases associated with NO imbalance.
[0008] Nitric oxide (NO), S-nitrosoglutathione (GSNO), and S-nitrosoglutathione reductase (GSNOR) regulate normal lung physiology and contribute to lung pathophysiology. Under normal conditions, NO and GSNO maintain normal lung physiology and function via their anti-inflammatory and bronchodilatory actions. Lowered levels of these mediators in pulmonary diseases such as asthma, chronic obstructive pulmonary disease (COPD) may occur via up-regulation of GSNOR enzyme activity. These lowered levels of NO and GSNO, and thus lowered anti-inflammatory capabilities, are key events that contribute to pulmonary diseases and which can potentially be reversed via GSNOR inhibition.
[0009] S-nitrosoglutathione (GSNO) has been shown to promote repair and/or regeneration of mammalian organs, such as the heart (Lima et al., 2010), blood vessels (Lima et al., 2010) skin (Georgii et al., 2010), eye or ocular structures (Haq et al., 2007) and liver (Prince et al., 2010). S-nitrosoglutathione reductase (GSNOR) is the major catabolic enzyme of GSNO. Inhibition of GSNOR is thought to increase endogenous GSNO.
[0010] Inflammatory bowel diseases (IBD’s), including Crohn’s and ulcerative colitis, are chronic inflammatory disorders of the gastrointestinal (GI) tract, in which NO, GSNO, and GSNOR can exert influences. Under normal conditions, NO and GSNO function to maintain normal intestinal physiology via anti-inflammatory actions and maintenance of the intestinal epithelial cell barrier. In IBD, reduced levels of GSNO and NO are evident and likely occur via up-regulation of GSNOR activity. The lowered levels of these mediators contribute to the pathophysiology of IBD via disruption of the epithelial barrier via dysregulation of proteins involved in maintaining epithelial tight junctions. This epithelial barrier dysfunction, with the ensuing entry of micro-organisms from the lumen, and the overall lowered anti-inflammatory capabilities in the presence of lowered NO and GSNO, are key events in IBD progression that can be potentially influenced by targeting GSNOR.
[0011] Cell death is the crucial event leading to clinical manifestation of
hepatotoxicity from drugs, viruses and alcohol. Glutathione (GSH) is the most abundant redox molecule in cells and thus the most important determinant of cellular redox status. Thiols in proteins undergo a wide range of reversible redox modifications during times of exposure to reactive oxygen and reactive nitrogen species, which can affect protein activity. The maintenance of hepatic GSH is a dynamic process achieved by a balance between rates of GSH synthesis, GSH and GSSG efflux, GSH reactions with reactive oxygen species and reactive nitrogen species and utilization by GSH peroxidase. Both GSNO and GSNOR play roles in the regulation of protein redox status by GSH.
[0012] Acetaminophen overdoses are the leading cause of acute liver failure (ALF) in the United States, Great Britain and most of Europe. More than 100,000 calls to the U.S. Poison Control Centers, 56,000 emergency room visits, 2600 hospitalizations, nearly 500 deaths are attributed to acetaminophen in this country annually. Approximately, 60% recover without needing a liver transplant, 9% are transplanted and 30% of patients succumb to the illness. The acetaminophen-related death rate exceeds by at least three-fold the number of deaths due to all other idiosyncratic drug reactions combined (Lee, Hepatol Res 2008; 38 (Suppl. 1):S3-S8).
[0013] Liver transplantation has become the primary treatment for patients with fulminant hepatic failure and end-stage chronic liver disease, as well as certain metabolic liver diseases. Thus, the demand for transplantation now greatly exceeds the availability of donor organs, it has been estimated that more than 18 000 patients are currently registered with the United Network for Organ Sharing (UNOS) and that an additional 9000 patients are added to the liver transplant waiting list each year, yet less than 5000 cadaveric donors are available for transplantation.
[0014] Currently, there is a great need in the art for diagnostics, prophylaxis, ameliorations, and treatments for medical conditions relating to increased NO synthesis and/or increased NO bioactivity. In addition, there is a significant need for novel compounds, compositions, and methods for preventing, ameliorating, or reversing other NO-associated disorders. The present invention satisfies these needs.
Schemes 1-6 below illustrate general methods for preparing analogs.
[00174] For a detailed example of General Scheme 1 see Compound IV-1 in Example 1.
[00175] For a detailed example of Scheme 2, A conditions, see Compound IV-2 in Example 2.
[00176] For a detailed example of Scheme 2, B conditions, see Compound IV-8 in Example 8.
[00177] For a detailed example of Scheme 3, see Compound IV-9 in Example 9.
[00178] For a detailed example of Scheme 4, Route A, see Compound IV-11 in Example 11.
[00179] For a detailed example of Scheme 4, Route B, see Compound IV-12 in Example 12.
[00180] For a detailed example of Scheme 5, Compound A, see Compound IV-33 in Example 33.
[00181] For a detailed example of Scheme 5, Compound B, see Compound IV-24 in Example 24.
[00182] For a detailed example of Scheme 5, Compound C, see Compound IV-23 in Example 23.
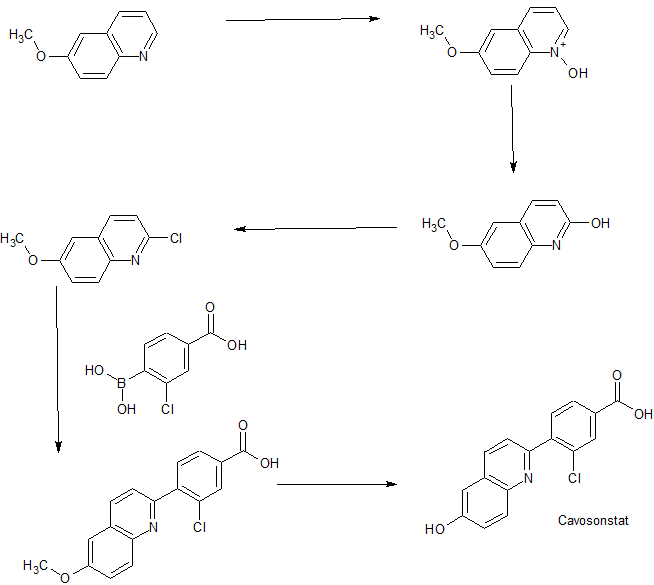
Example 8: Compound IV-8: 3-chloro-4-(6-hydroxyquinolin-2-yl)benzoic acid
[00209] Followed Scheme 2, B conditions:
[00210] Step 1: Synthesis of 3-chloro-4-(6-methoxyquinolin-2-yl)benzoic acid:
[00211] A mixture of 2-chloro-6-methoxyquinoline (Intermediate 1) (200 mg, 1.04 mmol), 4-carboxy-2-chlorophenylboronic acid (247 mg, 1.24 mmol) and K2CO3(369 mg, 2.70 mmol) in DEGME / H2O (7.0 mL / 2.0 mL) was degassed three times under N2 atmosphere. Then PdCl2(dppf) (75 mg, 0.104 mmol) was added and the mixture was heated to 110 °C for 3 hours under N2 atmosphere. The reaction mixture was diluted with EtOAc (100 mL) and filtered. The filtrate was washed with brine (20 mL), dried over Na2SO4, filtered and concentrated to give 3-chloro-4-(6-methoxyquinolin-2-yl)benzoic acid (150 mg, yield 46%) as a yellow solid, which was used for the next step without further purification.
[00212] Step 2: Synthesis of Compound IV-8: To a suspension of 3-chloro-4-(6-methoxyquinolin-2-yl)benzoic acid (150 mg, 0.479 mmol) in anhydrous CH2Cl2 (5 mL) was added AlCl3 (320 mg, 2.40 mmol). The reaction mixture was refluxed overnight. The mixture was quenched with saturated NH4Cl (10 mL) and the aqueous layer was extracted with CH2Cl2 / MeOH (v/v=10: l, 30 mL x3). The combined organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated to give the crude product, which was purified by prep-HPLC (0.1% TFA as additive) to give 3-chloro-4-(6-hydroxyquinolin-2-yl)benzoic acid (25 mg, yield 18%). 1H NMR (DMSO, 400 MHz): δ 10.20 (brs, 1H), 8.30 (d, J = 8.4 Hz, 1H), 8.10-8.00 (m, 2H), 7.95 (d, J = 9.2 Hz, 1H), 7.80 (d, J = 8.0 Hz, 1H), 7.72 (d, J = 8.8 Hz, 1H), 7.38 (dd, J = 6.4, 2.8 Hz, 1H), 7.22 (d, J = 2.4 Hz, 1H), MS (ESI): m/z 299.9 [M+H]+.
REFERENCES
1: Donaldson SH, Solomon GM, Zeitlin PL, Flume PA, Casey A, McCoy K, Zemanick ET,
Mandagere A, Troha JM, Shoemaker SA, Chmiel JF, Taylor-Cousar JL.
Pharmacokinetics and safety of cavosonstat (N91115) in healthy and cystic
fibrosis adults homozygous for F508DEL-CFTR. J Cyst Fibros. 2017 Feb 13. pii:
S1569-1993(17)30016-4. doi: 10.1016/j.jcf.2017.01.009. [Epub ahead of print]
PubMed PMID: 28209466.
//////////Cavosonstat, N-91115, Orphan Drug Status, NCT02589236, N91115-2CF-05, SNO-6, PHASE 2, N30 Pharma, Nivalis Therapeutics, CYSTIC FIBROSIS, FAST TRACK
O=C(O)C1=CC=C(C2=NC3=CC=C(O)C=C3C=C2)C(Cl)=C1