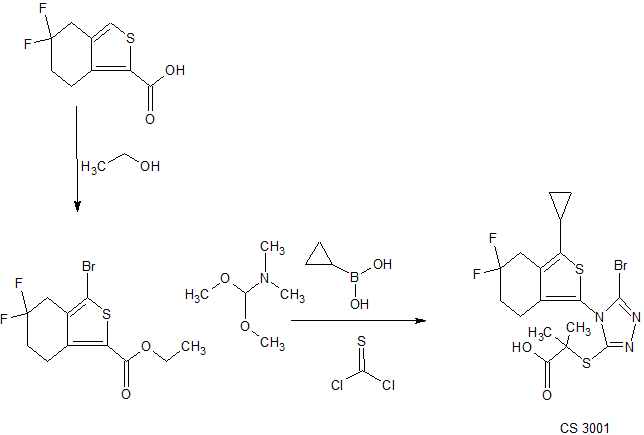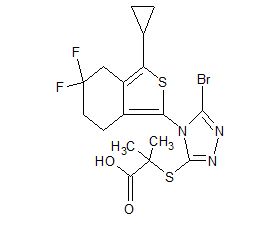
CS-3001
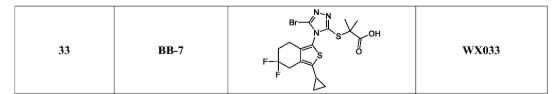
BB 7, VX 033
CAS 2159116-56-8
Propanoic acid, 2-[[5-bromo-4-(3-cyclopropyl-5,5-difluoro-4,5,6,7-tetrahydrobenzo[c]thien-1-yl)-4H-1,2,4-triazol-3-yl]thio]-2-methyl-
- Molecular Weight, 478.37
C17 H18 Br F2 N3 O2 S2
CStone Pharmaceuticals Co Ltd, JUNE 2018 IND FILED CHINA
URAT1 inhibitor – useful for treating hyperuricemia and gout.
The compound was originally claimed in WO2017202291 , covering thiophene derivative URAT1 inhibitors, useful for treating hyperuricemia and gouty arthritis, assigned to Medshine Discovery Inc , but naming the inventors.and has been reported in some instances to be a URAT1 modulator. In June 2018, an IND application was filed in
Uric acid is a product of the metabolism of terpenoids in animals. For humans, due to the lack of uric acid enzymes that continue to oxidatively degrade uric acid, uric acid is excreted in the human body as the final product of sputum metabolism through the intestines and kidneys. Renal excretion is the main pathway for uric acid excretion in humans. The upper limit of the normal range of uric acid concentration in the human body is: male 400 μmol/L (6.8 mg/dL) and female 360 μmol/L (6 mg/dL). Abnormal uric acid levels in the human body are often due to an increase in uric acid production or a decrease in uric acid excretion. Conditions associated with abnormal levels of uric acid include hyperuricemia, gout, and the like.
Hyperuricemia refers to a disorder in which the metabolism of substances in the human body is disordered, resulting in an increase or decrease in the synthesis of uric acid in the human body, and an abnormally high level of uric acid in the blood. Gouty arthritis refers to the fact that when uric acid is more than 7 mg/dL in human blood, uric acid is deposited as a monosodium salt in the joints, cartilage and kidneys, causing excessive reaction (sensitivity) to the body’s immune system and causing painful inflammation. The general site of attack is the big toe joint, ankle joint, knee joint and so on. Red, swollen, hot, and severe pain in the site of acute gout attacks, usually in the midnight episode, can make people wake up from sleep. In the early stages of gout, the attack is more common in the joints of the lower extremities. Hyperuricemia is the pathological basis of gouty arthritis. The use of drugs to lower blood uric acid concentration is one of the commonly used methods to prevent gouty arthritis.
In Europe and the United States, the onset of hyperuricemia and gout disease is on the rise. Epidemiological studies have shown that the incidence of gouty arthritis accounts for 1-2% of the total population and is the most important type of arthritis in adult males. Bloomberg estimates that there will be 17.7 million gout patients in 2021. In China, the survey showed that among the population aged 20 to 74, 25.3% of the population had a high blood uric acid content and 0.36% had gout disease. At present, clinical treatment drugs mainly include 1) inhibition of uric acid-producing drugs, such as xanthine oxidase inhibitor allopurinol and febuxostat; 2) uric acid excretion drugs, such as probenecid and benzbromarone; 3) Inflammation inhibitors, such as colchicine. These drugs have certain defects in treatment, poor efficacy, large side effects, and high cost are some of the main bottlenecks in their clinical application. It has been reported that 40%-70% of patients with serum uric acid levels do not meet the expected therapeutic goals (<6mg/dL) after receiving standard treatment.
As a uric acid excretion agent, its mechanism of action is to reduce the reabsorption of uric acid by inhibiting the URAT1 transporter on the brush-like edge membrane of the proximal convoluted tubule. Uric acid is a metabolite of sputum in the body. It is mainly filtered by glomerulus in the original form, reabsorbed and re-secreted by the renal tubules, and finally excreted through the urine. Very few parts can be secreted into the intestinal lumen by mesenteric cells. The S1 segment of the proximal convoluted tubule is a site of uric acid reabsorption, and 98% to 100% of the filtered uric acid enters the epithelial cells through the uric acid transporter URAT1 and the organic anion transporter OAT4 on the brush epithelial cell border of the tubular epithelial cells. The uric acid entering the epithelial cells is reabsorbed into the capillaries around the tubules via the renal tubular basement membrane. The S2 segment of the proximal convoluted tubule is the site of re-secretion of uric acid, and the amount secreted is about 50% of the excess of the small filter. The uric acid in the renal interstitial enters the epithelial cells first through the anion transporters OAT1 and OAT3 on the basal membrane of the tubular epithelial cells. The uric acid entering the epithelial cells passes through another anion transporter MRP4 on the brush border membrane and is discharged into the small lumen. The S3 segment of the proximal convoluted tubule may be a reabsorption site after uric acid secretion, and the amount of reabsorption is about 40% of the excess of the microsphere filtration, and similar to the first step of reabsorption, URAT1 may be a key reabsorption transporter. Therefore, if the urate transporter URAT1 can be significantly inhibited, it will enhance the excretion of uric acid in the body, thereby lowering blood uric acid level and reducing the possibility of gout attack.
In December 2015, the US FDA approved the first URAT1 inhibitor, Zurampic (Leinurad). The 200 mg dose was approved in combination with xanthine oxidase inhibitor XOI (such as Febuxostat, etc.) for the treatment of hyperuricemia and gouty arthritis, but the combination was compared with the xanthine oxidase inhibitor alone. The effect is not very significant. The Zurampic 400 mg dose was not approved due to significant toxic side effects at high doses (the incidence of renal-related adverse events, especially the incidence of kidney stones). Therefore, the FDA requires the Zurampic label to be filled with a black box warning to warn medical staff Zulampic of the risk of acute kidney failure, especially if it is not used in conjunction with XOI. If the over-approved dose uses Zurampic, the risk of kidney failure is even greater. high. At the same time, after the FDA asked for the listing of Zurampic, AstraZeneca continued its investigation of kidney and cardiovascular safety. Therefore, the development of a new type of safe blood-supplemented uric acid drug has become a strong demand in this field.
WO2009070740 discloses Leinurad, which has the following structure:
PATENT
WO-2019101058
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019101058&tab=FULLTEXT&maxRec=1000
Novel crystalline forms of URAT1 inhibitor (designated as Forms A and B) are claimed. The compounds are disclosed to be useful for treating hyperuricemia and gouty arthritis.
Novel crystalline forms of a URAT1 inhibitor, designated as Forms A and B, and their preparation.
Example 1: Preparation of a compound of formula (I)
Step 1: Synthesis of Compound 2
In a three-necked flask (10 L), 4.5 L of dimethyl sulfoxide was added, and potassium t-butoxide (836.66 g, 7.46 mol, 2 eq) was added with stirring, and stirring was continued for 10 minutes until the dissolution was clear, and then cooled to an ice water bath. The internal temperature of the reaction solution was 20-25 °C. To the above solution, a solution of Compound 1 (500.05 g, 3.73 mol, 1 eq) in dimethyl sulfoxide (500 mL) was added dropwise, and the mixture was stirred for 30 minutes, and then carbon disulfide (283.86 g, 3.73 mol, 1 eq) was added dropwise thereto. ), after the completion of the dropwise addition, the reaction was stirred for 30 minutes. Further, ethyl bromoacetate (1250 g, 7.46 mol, 2 eq) was added dropwise thereto, and the mixture was stirred for further 2 hours. Finally, potassium carbonate (515.52 g, 7.46 mol, 1 eq) was added, and the temperature was raised to an internal temperature of 65 ° C, and the reaction was further stirred for 8 hours. After the reaction was completed, the reaction solution was cooled to room temperature. The reaction solution was diluted with ethyl acetate (10 L), and then 1M hydrochloric acid (2 L) and water (2 L) were added and stirred for 10 minutes, and the mixture was allowed to stand. The aqueous layer was separated and the organic phase was washed with water (2L×3). The combined aqueous layers were extracted with ethyl acetate (3L). All organic phases were combined and washed with saturated brine (2 L×2). The organic phase was dried over an appropriate amount of anhydrous sodium sulfate, and then filtered, and then evaporated. On the same scale, 6 batches were fed in parallel, and the combined black and red oily products were obtained. After the crude product was allowed to stand for 72 hours, a large amount of solid was precipitated, ethanol (2 L) was added thereto, stirred for 30 minutes, filtered, and the cake was collected and dried in vacuo to give Compound 2. . 1 H NMR (400 MHz, CDCl3 . 3 ) [delta]: 4.32 (Q, J = 7.2 Hz, 2H), 4.19 (Q, J = 7.2 Hz, 2H), 3.56 (S, 2H), 3.25 (T, J = 6.8Hz , 2H), 3.19 (t, J = 14.4 Hz, 2H), 2.26-2.17 (m, 2H), 1.37 (t, J = 7.2 Hz, 3H), 1.27 (t, J = 7.2 Hz, 3H); MS m/z = 364.8 [M+H] + .
Step 2: Synthesis of Compound 3
Compound 2 (241.00 g, 0.66 mol) was dissolved in ethanol (1 L) and placed in an autoclave (5 L), and Raney nickel (120 g) was added under argon atmosphere, followed by the addition of ethanol (2 L). The autoclave was charged and replaced with argon three times, then replaced with hydrogen three times, hydrogen was charged to a pressure of 2.0 MP in the autoclave, stirred and heated to an internal temperature of 85 ° C for 28 hours. The reaction was stopped, the reaction system was cooled to room temperature, the reaction solution was filtered, and the filter cake was washed three times with ethanol, 0.5 L each time. The filtrates were combined and then dried to give compound 3. . 1 H NMR (400 MHz, CDCl3 . 3 ) [delta]: 7.09 (S, IH), 4.26 (Q, J = 7.2 Hz, 2H), 3.20 (T, J = 6.8Hz, 2H), 3.12 (T, J = 14.4Hz , 2H), 2.20-2.10 (m, 2H), 1.30 (t, J = 6.8 Hz, 3H); MS m/z = 247.0 [M+H] + .
Step 3: Synthesis of Compound 4
Compound 3 (406.2 g, 1.65 mol, 1 eq) was dissolved in acetonitrile (6 L), then N-bromosuccinimide (1484.2 g, 6.60 mol, 4 eq) was slowly added, and the obtained reaction mixture was at 23 to 25 ° C. The reaction was stirred for 12 hours. After the reaction was completed, the reaction liquid was concentrated to about 1.0 L. The solid was removed by filtration, and a saturated solution of sodium hydrogensulfite (1 L) was added to the filtrate and stirred for 10 min. Add acid ethyl ester and extract three times, 2L each time. The organic phases were combined and dried over anhydrous sodium sulfate. The desiccant was removed by filtration, and the filtrate was concentrated under reduced pressure. Petroleum ether (3 L) was added to the residue, and the mixture was stirred at 30 ° C for 30 minutes. After filtration, the filter cake was washed 5 times with petroleum ether, 200 mL each time, until no product remained in the filter cake. Combine all the organic phases and spin dry to obtain a crude product. Petroleum ether (100 mL) was added to the crude product, stirred well, filtered, and filtered, and then dried in vacuo. . 1 H NMR (400 MHz, CDCl3 . 3) [delta]: 4.24 (Q, J = 7.2 Hz, 2H), 3.19 (T, J = 6.8Hz, 2H), 2.95 (T, J = 14.4Hz, 2H), 2.17-2.07 (m, 2H), 1.29 (t, J = 7.2 Hz, 3H).
Step 4: Synthesis of Compound 5
Compound 4 (340.21 g, 1.05 mol), cyclopropylboronic acid (108.12 g, 1.26 mol), anhydrous potassium phosphate (444.98 g, 2.10 mol), palladium acetate (12.03 g, 53.58 mmol) and 2-dicyclohexyl Phospho-2′,4′,6′-triisopropylbiphenyl (23.86 g, 50.05 mmol) was added to a mixed solvent of toluene and water (10:1, 3.4 L/340 mL), and the reaction flask was replaced with nitrogen. After that, place it in an oil bath. The reaction solution was heated at an internal temperature of 80 ° C, and the reaction was stirred at this temperature for 16 hours. After completion of the reaction, the reaction solution was cooled to room temperature, and tris-thiocyanic acid (6.51 g, suspended in ethanol (34 mL)) was added to the reaction mixture and stirred for 0.5 hour. On a similar scale (300.00 g of compound 4), 5 batches were fed in parallel and combined. After filtration, the organic phase was separated and the aqueous phase was extracted with ethyl acetate (250mL). The organic phases were combined and dried over anhydrous sodium sulfate. The desiccant was removed by filtration, and the filtrate was concentrated under reduced pressure to yield crude crude oil. After the crude product was allowed to stand for 20 hours, a yellow solid was precipitated, and petroleum ether (3 L) was added thereto and stirred for 1 hour. Filtration and drying of the filter cake in vacuo gave compound 5. . 1 H NMR (400 MHz, CDCl3 . 3 ) [delta]: 4.29 (Q, J = 7.2 Hz, 2H), 3.23 (T, J = 6.4Hz, 2H), 3.16 (T, J = 14.8 Hz, 2H), 2.24-2.18 (m, 2H), 1.95-1.85 (m, 1H), 1.35 (t, J = 6.8 Hz, 3H), 1.09-1.07 (m, 2H), 0.77-0.75 (m, 2H).
Step 5: Synthesis of Compound 6
Compound 5 (619.27 g, 2.16 mol) was added to a mixed solution of ethanol and water (3 L/3 L) of sodium hydroxide (173.55 g, 4.33 mol), and the reaction liquid was heated to an internal temperature of 60 ° C to stir the reaction 3 hour. After the reaction was completed, the reaction solution was cooled to room temperature. On a similar scale (750.17 g of compound 5), 1 batch was fed in parallel and combined. The combined reaction solution was extracted with petroleum ether (4 L). The organic phase was separated and the organic phase was backwashed twice with water (1.5L x 2). The aqueous phases were combined and concentrated under reduced pressure to remove ethanol. Water was added to the aqueous phase to dilute to 13 L, and then slowly added with dilute hydrochloric acid (3 M) to adjust to pH = 2, and a large amount of pale yellow solid precipitated. Filter and filter cake with water (3.0L x 2). After draining, the filter cake was collected and dried under vacuum at 60 ° C to give Compound 6. . 1 H NMR (400 MHz, DMSO-D . 6 ) [delta]: 12.79 (brs, IH), 3.23 (T, J = 14.8 Hz, 2H), 3.07 (T, J = 6.8Hz, 2H), 2.27-2.20 (m, 2H), 2.19-2.02 (m, 1H), 1.09-1.04 (m, 2H), 0.68-0.66 (m, 2H).
Step 6: Synthesis of Compound 7
Compound 6 (641.27 g, 2.48 mol), triethylamine (754.07 g, 7.45 mol) and diphenyl azide (1025.34 g, 3.73 mol) were added to t-butanol (6.5 L) with stirring. The reaction solution was heated in a 100 ° C oil bath for 16 hours. After the reaction was completed, it was cooled to room temperature. On a similar scale (650.00 g of compound 6), 4 batches were fed in parallel and combined. The reaction mixture was combined and concentrated under reduced pressure to remove t-butyl alcohol. The remaining black residue was dissolved with ethyl acetate (10L). Dry with an appropriate amount of anhydrous sodium sulfate. The desiccant was removed by filtration, and the filtrate was concentrated under reduced pressure to give a crude brown solid. Petroleum ether (8 L) was added to the crude product and stirred for 2 hours. After filtration, the filter cake was rinsed with petroleum ether (1 L) in portions, and the filter cake was vacuum dried in a vacuum oven at 60 ° C to obtain Compound 7. . 1 H NMR (400 MHz, CDCl3 . 3 ) [delta]: 6.31 (brs, IH), 3.11 (T, J = 14.8 Hz, 2H), 2.66 (T, J = 6.8Hz, 2H), 2.23-2.15 (m, 2H) , 1.82-1.75 (m, 1H), 1.51 (s, 9H), 0.94-0.90 (m, 2H), 0.68-0.65 (m, 2H).
Step 7: Synthesis of Compound 8
Compound 7 (1199.17 g, 3.64 mol) was added to ethyl acetate (2 L), and then stirred and then ethyl acetate (4L, 16. The reaction solution was reacted at 15 ° C for 2.5 hours, and then placed in a 40 ° C warm water bath to continue the reaction for 2 hours. After the reaction was completed, a large amount of dark red solid precipitated. Filter and filter cake was rinsed with ethyl acetate (2.0 L). The filter cake was dried under vacuum in a vacuum oven at 60 ° C to give compound 8. . 1 H NMR (400 MHz, DMSO-D . 6 ) [delta]: 3.17 (T, J = 14.8 Hz, 2H), 2.82 (T, J = 6.8Hz, 2H), 2.25-2.15 (m, 2H), 2.00-1.94 ( m, 1H), 0.99-0.95 (m, 2H), 0.58-0.54 (m, 2H); MS m/z = 229.8 [M+H-HCl] + .
Step 8: Synthesis of Compound 9
In a 3 L three-necked flask, Compound 8 (301.25 g) was added to tetrahydrofuran (600 mL), and the mixture was cooled to an internal temperature of 0 to 10 ° C under ice-cooling. Diisopropylethylamine (635.72 g) was added dropwise, and after completion of the dropwise addition, the ice water bath was removed, and the mixture was stirred at an internal temperature of 10 to 15 ° C for about 10 minutes. Filter and filter cake was washed with tetrahydrofuran (100 mL x 2). The filtrates were combined to give a solution A for use.
Tetrahydrofuran (2 L) was added to a 5 L reaction flask containing thiophosgene (257.48 g). The mixture was stirred and cooled to an internal temperature of 0 to 10 ° C in an ice water bath, and the solution A was slowly added dropwise thereto, and the dropwise addition was completed within about 5.5 hours, and stirring was continued for 10 minutes. After the reaction was completed, it was filtered, and the filter cake was washed with tetrahydrofuran (150 mL × 2). The filtrate was combined and concentrated under reduced pressure to remove solvent. Tetrahydrofuran (400 mL) was added to the residue, which was dissolved to give a solution B.
The hydrazine hydrate (112.94 g) was added to tetrahydrofuran (2.5 L), and the mixture was cooled to an internal temperature of 5 to 10 ° C under ice-cooling. Solution B was added dropwise, and the addition was completed for about 2 hours, and stirring was continued for 10 minutes. After the reaction was completed, the reaction was stopped. The ice water bath was removed, N,N-dimethylformamide dimethyl acetal (333.45 g) was added, and the mixture was heated to an internal temperature of 60 to 65 ° C, and the reaction was stopped after the heat retention reaction for 3 hours.
The reaction solution was dried to dryness, and ethyl acetate (2 L) and purified water (1L) were added to the residue, and the mixture was stirred. The pH was adjusted to 5-6 with 10% hydrobromic acid, stirring was continued for 5 minutes, and allowed to stand for 10 minutes. Dispense and separate the aqueous phase. The organic phase was washed with pure water (500 mL x 2). The combined aqueous phases were extracted with EtOAc (1 mL). The desiccant was removed by filtration, and the filtrate was concentrated to dryness to dryness. n-Heptane (2.0 L) and tert-butyl methyl ether (150 mL) were added to the crude product, and the mixture was stirred ( stirring speed 550 rpm) for 18 hours. Filter and filter cake was washed with n-heptane (150 mL). The filter cake was collected and the filter cake was dried under vacuum at 60 ° C to give compound 9. . 1 H NMR (400 MHz, CDCl3 . 3 ) [delta]: 7.82 (S, IH), 3.20 (T, J = 14.8 Hz, 2H), 2.74 (T, J = 6.8Hz, 2H), 2.28-2.10 (m, 2H) , 1.98-1.82 (m, 1H), 1.06-1.02 (m, 2H), 0.75-0.71 (m, 2H); MS m/z = 313.9 [M+H] + .
Step 9: Synthesis of Compound 10
Acetonitrile (3 L) was placed in a 5 L three-necked flask. Compound 9 (303.25 g) and potassium carbonate (261.83 g) were added first with stirring. Further, methyl 2-bromoisobutyrate (203.85 g) was added, and the reaction system was replaced with nitrogen, and then heated to an internal temperature of 60 to 65 ° C, and the reaction was kept for about 2 hours. After the completion of the reaction, the heating was stopped, and the mixture was naturally cooled to 15 to 20 ° C under stirring. Filter and filter cake was washed with ethyl acetate (100 mL x 3). The filtrate was combined and concentrated under reduced pressure to dryness. The crude product was purified by column chromatography (mobile phase: ethyl acetate / n-heptane = 1:5 to 2:1). . 1 H NMR (400 MHz, CDCl3 . 3 ) [delta]: 8.20 (S, IH), 3.68 (S, 3H), 3.19 (T, J = 14.4Hz, 2H), 2.57 (T, J = 6.8Hz, 2H), 2.22 -2.12 (m, 2H), 1.93-1.83 (m, 1H), 1.67 (s, 6H), 1.08-1.03 (m, 2H), 0.73-0.69 (m, 2H); MS m/z = 414.0 [M +H] + .
Step 10: Synthesis of Compound 11
Acetonitrile (3.17 L) was placed in a 5 L three-necked flask. Under stirring, compound 10 (317.22 g) and thiocarbonyldiimidazole (26.94 g) were added, and the mixture was stirred at 16 to 20 ° C for 5 minutes. N-bromosuccinimide (158.60 g) was added and stirred for about 30 minutes with heat. After the reaction was over, the reaction was stopped. Filtration and concentration of the filtrate under reduced pressure afforded crude crude. The crude product was purified by column chromatography (EtOAc:EtOAc:EtOAc This crude product was dissolved in ethyl acetate (3.50 L) and washed with purified water (700 mL×4). The organic phase was separated and the organic phase was dried over anhydrous sodium sulfate. The desiccant was removed by filtration, and the filtrate was concentrated to dryness to give Compound 11. . 1 H NMR (400 MHz, CDCl3 . 3 ) [delta]: 3.73 (S, 3H), 3.22 (T, J = 14.4Hz, 2H), 2.53 (T, J = 6.8Hz, 2H), 2.24-2.14 (m, 2H) , 1.95-1.91 (m, 1H), 1.71 (d, J = 4.4 Hz, 6H), 1.11-1.07 (m, 2H), 0.78-0.74 (m, 2H); MS m/z = 491.7 [M+H ] + ,493.7[M+H+2] + .
Step 11: Synthesis of a compound of formula (I)
Tetrahydrofuran (1.2 L) was added to a 5 L reaction flask, and Compound 11 (243.03 g) was added with stirring. After the solution was dissolved, pure water (1.2 L) was added, and then lithium hydroxide monohydrate (125.46 g) was added, and the mixture was stirred at 20 to 25 ° C for about 2.5 hours. After the reaction was completed, the reaction was stopped. The reaction solution was concentrated under reduced pressure at 40 ° C to remove organic solvent. Pure water (1 L) was added to the residue, and the mixture was extracted with t-butyl methyl ether (300 mL). The aqueous phase was placed in a 10 L three-necked flask and cooled to 5 to 10 ° C in an ice bath. The pH was adjusted to 2 to 3 with a 40% hydrobromic acid solution, and a large amount of a pale yellow solid precipitated. Stirring was continued for 30 minutes, and the pH was again measured to be 2-3. Stirring was continued for 20 minutes and filtered. The filter cake was washed with pure water (150 mL x 3). The filter cake was collected, pure water (1500 mL) was added, and the mixture was beaten at room temperature for 1 hour. After filtration, the filter cake was washed with pure water (150 mL × 2), and the filter cake was collected and dried under vacuum at 40 ° C for 3 hours to obtain a compound of the formula (I). . 1 H NMR (400 MHz, the CD . 3 the OD) [delta]: 3.27 (T, J = 15.6Hz, 2H), 2.60-2.47 (m, 2H), 2.27-2.17 (m, 2H), 2.10-2.03 (m, IH) , 1.68 (d, J = 1.2 Hz, 6H), 1.15.10.10 (m, 2H), 0.80-0.71 (m, 2H); MS m/z = 477.99 [M+H] + , 480.1 [M+H+ 2] + .
Example 2: Preparation of Form A of Compound of Formula (I)
The compound of the formula (I) (50 mg) was added to a glass bottle, and methanol (0.4 mL) was added thereto, followed by stirring to a suspension or a solution. The suspension sample was placed in a thermomixer (40 ° C), shaken at 40 ° C for 60 hours, and then centrifuged to collect a sample. The above-mentioned lysed sample was volatilized at room temperature, centrifuged, and the sample was collected. The above sample was dried in a vacuum oven (40 ° C) overnight, and its crystalline form was examined by XRPD to obtain a crystal form of the final product having a crystalline form of the compound of the formula (I).
The compound of the formula (I) (50 mg) was added to a glass bottle, and ethyl acetate (0.4 mL) was added and stirred to a suspension or a solution. The suspension sample was placed in a thermomixer (40 ° C), shaken at 40 ° C for 60 hours, and then centrifuged to collect a sample. The above-mentioned lysed sample was volatilized at room temperature, centrifuged, and the sample was collected. The above sample was dried in a vacuum oven (40 ° C) overnight, and its crystalline form was examined by XRPD to obtain a crystal form of the final product having a crystalline form of the compound of the formula (I).
Example 3: Preparation of Form B of Compound of Formula (I)
The compound of the formula (I) (50 mg) was added to a glass bottle, tetrahydrofuran (0.4 mL) was added, and the mixture was stirred to dissolve. The above-mentioned lysed sample was volatilized at room temperature, centrifuged, and the sample was collected. The collected sample was dried in a vacuum oven (40 ° C) overnight, and its crystalline form was examined by XRPD to obtain a crystalline form of the final product in the form of Form B of the compound of formula (I).
Example 4: Solubility test of Form A of the compound of formula (I)
1. Preparation of diluent and mobile phase
Diluent: Accurately measure 300mL of pure water and 100mL of pure acetonitrile, mix in a 1L glass bottle, ultrasonic degassing for 10 minutes and then set aside.
Mobile phase A: 0.1% phosphoric acid aqueous solution
For example, remove 2.0 mL of phosphoric acid into 2000 mL of water, sonicate for 10 minutes, mix, and let cool to room temperature as mobile phase A.
Mobile phase B: acetonitrile.
2. Preparation of the reference solution (using the A crystal form itself as a control sample)
Accurately weigh 5 mg of Form A, place it in a sample vial, add 10 mL of diluent, sonicate for 5 minutes, then cool to room temperature and mix well, and mark it as working reference solution STD-1.
Accurately weigh 5 mg of Form A, place it in a sample vial, add 10 mL of diluent, sonicate for 5 minutes, then cool to room temperature and mix well, and mark it as working reference solution STD-2.
3. Preparation of linear solution
The above working reference solution STD-1 was diluted 1 time, 10 times, 100 times, 1000 times and 2000 times, and recorded as linear solutions L1, L2, L3, L4 and L5.
Accurately weigh 6mg of A crystal form into 8mL glass bottle, then accurately add 3mL different solvent (0.1N hydrochloric acid solution, 0.01N hydrochloric acid solution, purified water, pH3.8 buffer solution, pH4.5 buffer solution, pH5 .5 buffer solution, pH 6.0 buffer solution, pH 7.4 buffer solution, pH 6.8 buffer solution), made into a suspension. A stir bar was added to the above suspension, and the mixture was thoroughly stirred at 37 ° C in the dark. After stirring, the solids in the pH 7.4 buffer solution and the pH 6.8 buffer solution were all dissolved, and 6 mg of the A crystal form was accurately weighed, added to the buffer solution, and thoroughly stirred again to prepare a suspension. After stirring for 4 hours and 24 hours, the sample was centrifuged, and the solution was filtered through a filter and the concentration thereof was measured by HPLC. The HPLC analysis method is shown in Table 3.
Table 3: HPLC analysis methods
////////////CS-3001, BB 7, VX 033, CHINA, PRECLINICAL, CStone Pharmaceuticals, URAT1 inhibitor, hyperuricemia, gout
O=C(O)C(C)(C)Sc4nnc(Br)n4c2sc(c1CC(F)(F)CCc12)C3CC3