















ELAGOLIX
- Molecular FormulaC32H30F5N3O5
- Average mass631.590 Da
GNRH antagonist, Endometriosis
Endometriosis PREREGISTERED
Phase III Uterine leiomyoma
| Inventors | Yun-Fei Zhu, Chen Chen, Fabio C. Tucci, Zhiqiang Guo, Timothy D. Gross, Martin Rowbottom, R. Scott Struthers, |
| Applicant | Neurocrine Biosciences, Inc. |
WO 2005007165 PDT PATENT

| Inventors | Zhiqiang Guo, Yongsheng Chen, Dongpei Wu, Chen Chen, Warren Wade, Wesley J. Dwight, Charles Q. Huang, Fabio C. Tucci, |
| Applicant | Neurocrine Biosciences, Inc. |
- Originator Icahn School of Medicine at Mount Sinai
- Developer AbbVie; Neurocrine Biosciences
- Class Antineoplastics; Fluorinated hydrocarbons; Pyrimidines; Small molecules
- Mechanism of Action LHRH receptor antagonists
- Highest Development Phases
- Preregistration Endometriosis
- Phase III Uterine leiomyoma
- Discontinued Benign prostatic hyperplasia; Prostate cancer
- Most Recent Events
- 23 Nov 2017 AbbVie plans a phase III trial for Endometriosis (Monotherapy, Combination therapy) in USA in November 2017 (NCT03343067)
- 01 Nov 2017 Updated efficacy and adverse events data from two phase III extension trials in Endometriosis released by AbbVie
- 27 Oct 2017 Elagolix receives priority review status for Endometriosis in USA
SYN
Elagolix is a specific highly potent non-peptide, orally active antagonist of the GnRH receptor. This compound inhibits pituitary luteinizing hormone (LH) secretion directly, potentially preventing the several week delay and flare associated with peptide agonist therapy.

In 2010, elagolix sodium was licensed to Abbott by Neurocrine Biosciences for worldwide development and commercialization for the treatment of endometriosis. In January 2013, Abbott spun-off its research-based pharmaceutical business into a newly-formed company AbbVie.
AbbVie , following its spin-out from Abbott in January 2013, under license from Neurocrine , is developing elagolix, the lead from a series of non-peptide gonadotropin-releasing hormone antagonists, for treating hormone-dependent diseases, primarily endometriosis and uterine fibroids.
Elagolix sodium is an oral gonadotropin releasing hormone (GnRH) antagonist in development at Neurocrine Biosciences and Abbvie (previously Abbott). In 2017, Abbvie submitted a New Drug Application (NDA) in the U.S. for the management of endometriosis with associated pain. The candidate is being evaluated in phase III trials for the treatment of uterine fibroids.
Elagolix (INN, USAN) (former developmental code names NBI-56418, ABT-620) is a highly potent, selective, orally-active, short-duration, non-peptide antagonist of the gonadotropin-releasing hormone receptor (GnRHR) (KD = 54 pM) which is under development for clinical use by Neurocrine Biosciences and AbbVie.[2][3] As of 2017, it is in pre-registration for the treatment of endometriosis and phase III clinical trials for the treatment of uterine leiomyoma.[1][4] The drug was also under investigation for the treatment of prostate cancer and benign prostatic hyperplasia, but development for these indications was ultimately not pursued.[4] Elagolix is the first of a new class of GnRH inhibitors that have been denoted as “second-generation”, due to their non-peptide nature and oral bioavailability.[1]
Because of the relatively short elimination half-life of elagolix, the actions of gonadotropin-releasing hormone (GnRH) are not fully blocked throughout the day.[1][5] For this reason, gonadotropin and sex hormone levels are only partially suppressed, and the degree of suppression can be dose-dependently adjusted as desired.[1][5] In addition, if elagolix is discontinued, its effects are rapidly reversible.[1][5] Due to the suppression of estrogen levels by elagolix being incomplete, effects on bone mineral density are minimal, which is in contrast to first-generation GnRH inhibitors.[6][7] Moreover, the incidence and severity of menopausal side effects such as hot flashes are also reduced relative to first-generation GnRH inhibitors.[1][5]
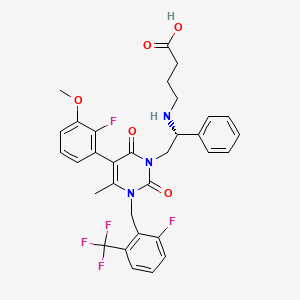
Elagolix sodium is a non-peptide antagonist of the gonadotropin-releasing hormone receptor and chemically known as sodium;4-[[(lR)-2-[5-(2-fluoro-3-methoxyphenyl)-3-[[2-fluoro-6-(trifluoromethyl)phenyl]methyl] -4-methyl-2,6-dioxopyrimidin- 1 -yl] -1 -phenylethyl] amino] butanoate as below.

The US patent number 7056927 B2 discloses, elagolix sodium salt as a white solid and process for its preparation in Example-1; Step-IH.
The US patent number 8765948 B2 discloses a process for preparation of amorphous elagolix sodium by spray drying method and solid dispersion of amorphous elagolix sodium with a polymer.
The US patent number 7056927 B2 discloses a process for preparation of elagolix sodium salt in Example -1 as given in below scheme -I.

Scheme -I
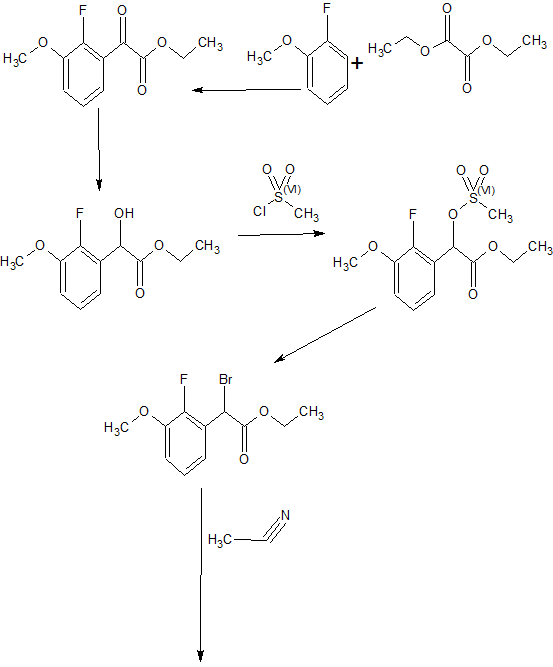
The US patent number 8765948 B2 describes a process for preparation of elagolix sodium in example- 1 and 4 as given below scheme-II:

(1c) (1e) (4a)
Scheme-II
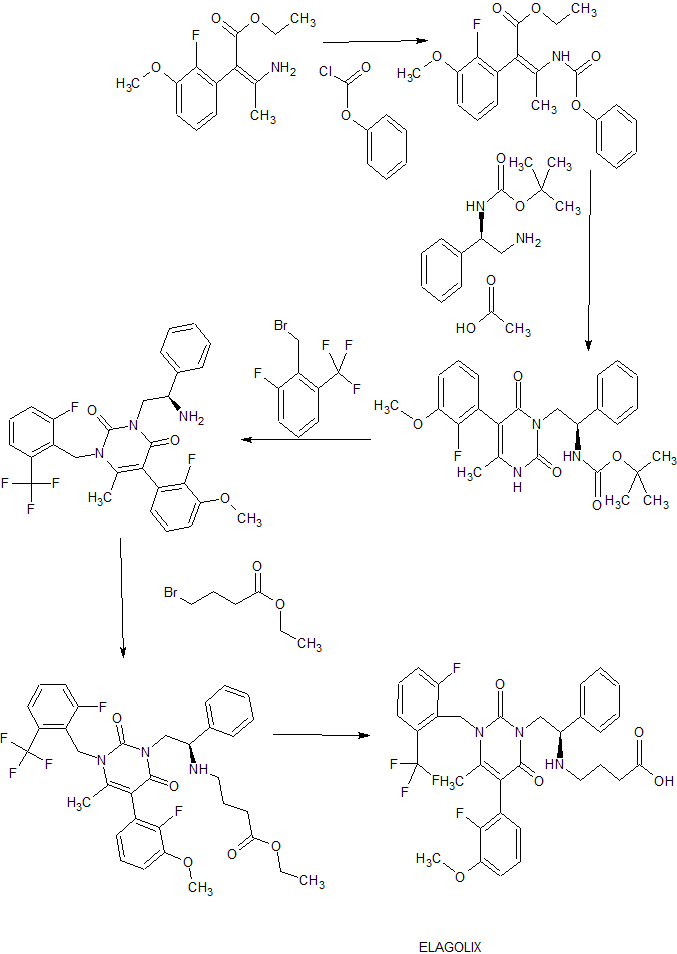
Further, the US patent number 8765948 B2 discloses an alternate process for the preparation of compound of formula (le) as mentioned below scheme-Ill.


Scheme -III
PATENT
| WO2001055119A2 * | Jan 25, 2001 | Aug 2, 2001 | Neurocrine Biosciences, Inc. | Gonadotropin-releasing hormone receptor antagonists and methods relating thereto |
PATENT
WO 2005007165
https://encrypted.google.com/patents/WO2005007165A1?cl=en
EXAMPLE 1
3-[2(R)-{HYD OXYCARBONYLPROPYL-AMINθ} -2-PHENYLETHYL]-5-(2-FLUORO-3- METHOXYPHENYL)-l-[2-FLUORO-6-(TRIFLUOROMETHYL)BENZYL]-6-METHYL- PYRIMIDINE-2,4(lH,3H)-DIONE

Step IA: Preparation of 2-fluoro-6-(trifluoromethyl)benzylamine la To 2-fluoro-6-(trifluoromethyl)benzonitrile (45 g, 0.238 mmol) in 60 mL of TΗF was added 1 M BΗ3:TΗF slowly at 60 °C and the resulting solution was refluxed overnight. The reaction mixture was cooled to ambient temperature. Methanol (420 mL) was added slowly and stirred well. The solvents were then evaporated and the residue was partitioned between EtOAc and water. The organic layer was dried over Na2SO4. Evaporation gave la as a yellow oil (46 g, 0.238 mmol). MS (C\) m/z 194.0 (MH+).
Step IB: Preparation of N-|“2-fluoro-6-(trifluoromethyl)benzyl‘|urea lb To 2-fluoro-6-(trifluoromethyl)benzylamine la (51.5 g, 0.267 mmol) in a flask, urea (64 g, 1.07 mmol), HC1 (cone, 30.9 mmol, 0.374 mmol) and water (111 mL) were added. The mixture was refluxed for 6 hours. The mixture was cooled to ambient temperature, further cooled with ice and filtered to give a yellow solid. Recrystallization with 400 mL of EtOAc gave lb as a white solid (46.2 g, 0J96 mmol). MS (CI) m/z 237.0 (MH+).
Step 1C: Preparation of l-[2-fluoro-6-(trifluoromethyl)benzyl]-6- methylpyrimidine-2.4(lH.3H)-dione lc Nal (43.9 g, 293 mmol) was added to N-[2-fluoro-6- (trifluoromethyl)benzyl]urea lb (46.2 g, 19.6 mmol) in 365 mL of acetonitrile. The resulting mixture was cooled in an ice-water bath. Diketene (22.5 mL, 293 mmol) was added slowly via dropping funnel followed by addition of TMSCl (37.2 mL, 293 mmol) in the same manner. The resulting yellow suspension was allowed to warm to room temperature slowly and was stirred for 20 hours. LC-MS showed the disappearance of starting material. To the yellow mixture 525 mL of water was added and stirred overnight. After another 20 hours stirring, the precipitate was filtered via Buchnner funnel and the yellow solid was washed with water and EtOAc to give lc as a white solid (48.5 g, 16 mmol). 1H ΝMR (CDC13) δ 2.15 (s, 3Η), 5.37 (s, 2H), 5.60 (s, 1H), 7.23-7.56 (m, 3H), 9.02 (s, 1H); MS (CI) m/z 303.0 (MH+).
Step ID: Preparation of 5-bromo-l -[2-fluoro-6-(trifluoromethyl)benzyl“|-6- methylpyrimidine-2.4(lH.3H)-dione Id Bromine (16.5 mL, 0.32 mmol) was added to l-[2-fluoro-6-
(trifluoromethyl)benzyl]-6-methylpyrimidine-2,4(lHJH)-dione lc (48.5 g, 0J6 mol) in 145 mL of acetic acid. The resulting mixture became clear then formed precipitate within an hour. After 2 hours stirring, the yellow solid was filtered and washed with cold EtOAc to an almost white solid. The filtrate was washed with sat. ΝaΗCO3 and dried over Na2SO4. Evaporation gave a yellow solid which was washed with EtOAC to give a light yellow solid. The two solids were combined to give 59.4 g of Id (0J56 mol) total. Η NMR (CDC13) δ 2.4 (s, 3H), 5.48 (s, 2H), 7.25-7.58 (m, 3H), 8.61 (s, 1H); MS (CI) m/z 380.9 (MH+). 5-Bromo-l-[2, 6-difluorobenzyl]-6-methylpyrimidine-2,4(lHJH)-dione ld.l was made using the same procedure.
Step IE: Preparation of 5-bromo-l -r2-fluoro-6-(trifluoromethyl)benzyll-6- methyl-3-[2(R)-tert-butoxycarbonylamino-2-phenylethyll-pyrimidine-2.4(lHJH)-dione le To 5-bromo- 1 -[2-fluoro-6-(trifluoromethyl)benzyl]-6-methylpyrimidine- 2,4(lHJH)-dione Id (15 g, 39.4 mmol) in 225 mL of TΗF were added N-t-Boc-D- phenylglycinol (11.7 g, 49.2 mmol) and triphenylphosphine (15.5 g, 59J mmol), followed by addition of di-tert-butyl azodicarboxylate (13.6 g, 59J mmol). The resulting yellow solution was stirred overnight. The volatiles were evaporated and the residue was purified by silica gel with 3:7 EtOAc Ηexane to give le as a white solid (23.6 g, 39.4 mmol). MS (CI) m/z 500.0 (MΗ+-Boc).
Step IF: Preparation of 3-[2(R)-amino-2-phenylethyll-5-(2-fluoro-3- methoxyphenyl)-l-[2-fluoro-6-(trifluoromethyl)benzyll-6-methyl-pyrimidine- 2.4(lH.3H)-dione If To 5-bromo-l-[2-fluoro-6-(trifluoromethyl)benzyl]-6-methyl-3-[2(R)- tert-butoxycarbonylamino-2-phenylethyl]-pyrimidine-2,4(lH,3H)-dione le (15 g, 25 mmol) in 30 mL/90 mL of Η2O/dioxane in a pressure tube were added 2-fluoro-3- methoxyphenylboronic acid (4.25 g, 25 mmol) and sodium carbonate (15.75 g, 150 mmol). N2 gas was bubbled through for 10 min.
Tetrakis(triphenylphosphine)palladium (2.9 g, 2.5 mmol) was added, the tube was sealed and the resulting mixture was heated with stirring at 90 °C overnight. After cooling to ambient temperature, the precipitate was removed by filtration. The volatiles were removed by evaporation and the residue was partitioned between EtOAc/sat. NaHCO3. The organic solvent was evaporated and the residue was chromatographed with 2:3 EtOAc/Hexane to give 13.4 g (20.8 mmol, 83 %) yellow solid. This yellow solid (6.9 g, 10.7 mmol) was dissolved in 20 mL/20 mL CH2C12/TFA. The resulting yellow solution was stirred at room temperature for 2 hours. The volatiles were evaporated and the residue was partitioned between EtOAc/ sat. NaHCO3. The organic phase was dried over Na2SO4. Evaporation gave If as a yellow oil (4.3 g, 7.9 mmol, 74%). Η NMR (CDC13) δ 2.03 (s, 3H), 3.72-4.59 (m, 6H), 5.32-5.61 (m, 2H), 6.74-7.56 (m, 11H); MS (CI) m/z 546.0 (MH+). 3-[2(R)-amino-2-phenylethyl]-5-(2-fluoro-3-methoxyphenyl)-l-[2,6- difluorobenzyl]-6-methyl-pyrimidine-2,4(lH,3H)-dione lf.l was made using the same procedure described in this example.
Step 1G: Preparation of 3-[2(R)- {ethoxycarbonylpropyl-amino} -2-phenylethyll-5-
(2-fluoro-3 -methoxyphenyl)- 1 -[2-fluoro-6-(trifluoromethyl)benzyl“|-6-methyl- pyrimidine-2,4(lHJH)-dione lg To compound 3-[2(R)-amino-2-phenylethyl]-5-(2-fluoro-3- methoxyphenyl)-l-[2-fluoro-6-(trifluoromethyl)benzyl]-6-methyl-pyrimidine- 2,4(lH,3H)-dione If (5 g, 9.4 mmol) in 100 mL of acetonitrile were added ethyl 4- bromobutyrate (4 mL, 28.2 mmol) and Ηunig’s base (1.6 mL, 9.4 mmol). After reflux at 95 °C overnight, the reaction mixture was cooled to ambient temperature and the volatiles were removed. The residue was chromatographed with 10:10: 1 EtOAc/Ηexane/Et3N to give lg as a yellow oil (3.0 g, 4.65 mmol). MS (CI) m/z 646.2 (MH+).
Step 1H: Preparation of 3-[2(R)- {hydroxycarbonylpropyl-amino} -2-phenylethyl]- 5-(2-fluoro-3-methoxyphenyl)-l- 2-fluoro-6-(trifluoromethyl)benzyl1-6-methyl- pyrimidine-2,4(lHJH)-dione 1-1 Compound 3-[2(R)- {ethoxycarbonylpropyl-amino} -2-phenylethyl]-5-(2- fluoro-3-methoxyphenyl)-l-[2-fluoro-6-(trifluoromethyl)benzyl]-6-methyl-pyrimidine- 2,4(lH,3H)-dione lg (2.6 g, 4.0 mmol) was dissolved in 30 mL/30 mL of TΗF/water. Solid NaOΗ (1.6 g, 40 mmol) was added and the resulting mixture was heated at 50 °C overnight. The mixture was cooled to ambient temperature and the volatiles were evaporated. Citric acid was added to the aqueous solution until pΗ = 3. Extraction with EtOAc followed by evaporation of solvent gave 1.96 g of a white gel. The gel was passed through a Dowex MSC-1 macroporous strong cation-exchange column to convert to sodium salt. Lyopholization gave white solid 1-1 as the sodium salt (1.58 g, 2.47 mmol). Η NMR (CD3OD) δ 1.69-1.77 (m, 2H), 2.09 (s, 3H), 2.09-2.19 (t, J = 7.35 Hz, 2H), 2.49-2.53 (t, J = 735 H, 2H), 3.88 (s, 3H), 4.15-4.32 (m, 3H), 5.36-5.52 (m, 2H), 6.60-7.63 (m, 1 IH); HPLC-MS (CI) m/z 632.2 (MH+), tR = 26.45, (method 5)
PATENT
WO 2017221144
Process for the preparation of elagolix sodium and its polymorph forms and intermediates is claimed. Represents first filing from Dr. Reddy’s Laboratories Limited and the inventors on this API.
n a seventh aspect, the present invention provides a process for preparation of compound of formula (VII)

(VII)
wherein R is alkyl such as methyl, ethyl, propyl, isopropyl and the like,
comprising;
a) reacting the compound of formula (II) with compound of formula (III) to obtain the compound of formula (IV)

wherein t-BOC is tertiary butoxycarbonyl group; R is as described above
b) reacting the compound of formula (IV) with the compound of formula (V) to obtain the compound formula (VI), and

c) N-deprotection of the compound of formula (VI) to obtain the compound of formula
(VII)

(VI) (VII)
The reaction of compound of formula (II) with compound of formula (III) to obtain the compound of formula (IV) is carried in the presence of triarylphosphine such as triphenyl phosphine and the like and azodicarboxylates such as diethyl azodicarboxylate, diisopropyl azodicarboxylate and di-tert-butyl azodicarboxylate (DIAD) and the like.
The seventh aspect of the present invention is depicted below scheme-IV.

Scheme-IV
The eighth aspect of the present invention is depicted below scheme-IV.

R=alkyl
Scheme-IV
Example 11: Preparation of ethyl (R)-4-((2-hydroxy-l-phenylethyl)amino)butanoate (Ilia; R is ethyl)
R-(-)-2-phenylglycinol (10 g), DMAP (0.17 g) were added in THF (80 ml) at room temperature under nitrogen atmosphere. Triethylamine (30.48 ml) was added to the reaction mixture and stirred for five minutes. Ethyl-4-bromo butyrate (15.64 ml) was added and the reaction mixture heated to 80°C then stirred for 16 hours. Water (20 volumes) followed by ethyl acetate (200 ml) were added to separate the aqueous and organic layer. The organic layer was washed with IN HC1 (100 ml) followed by neutralize the resulting aqueous layer with saturated sodium carbonate solution then extract with ethyl acetate (100 ml) and the organic layer was dried over anhydrous sodium sulfate then evaporated below 50°C under reduced pressure to obtain the title compound. Yield: 14.50 g. Purity: 94.75% (by HPLC). ¾ NMR (400 MHz, DMSO-d6): δ 7.17-7.30 (m, 5H), 4.83 (m, 1H), 3.99 (q, 2H), 3.58 (dd, 1H, J = 8.8, 4.4 Hz), 3.88 (m, 1H ), 3.27 (m, 1H), 2.38 (m, 1H), 2.26 (m, 3H), 2.10 (s, 1H), 1.61 (m, 2H), 1.12 (t, 3H); m/z: 252 (MH )
Example 12: Preparation of ethyl (R)-4-((tert-butoxycarbonyl)(2-hydroxy-l-phenylethyl) amino)butanoate (III; R is ethyl)
Ethyl (R)-4-((2-hydroxy-l-phenylethyl)amino)butanoate (14 g) was added to THF (140 ml) at room temperature. The reaction mixture was cooled to 0-5 °C. Triethylamine (16.9 mL) was added to the reaction mixture followed by Di-tert-butyl dicarbonate (13.37 g) was added to reaction mixture at 0-5 °C. The reaction mixture was heated to room temperature and stirred for 16 hours. Water (300 mL) and ethyl acetate (300 mL) were added and the layers were separated. The organic layer was washed with sodium chloride then died over sodium sulfate followed by evaporation at 45°C to obtain the crude compound. The crude compound was purified by silica gel (60/120 mesh) withl5-20% EtOAc/Hexane to obtain the title compound as a pale yellow syrup. Yield: 9.5 g. Purity: 95.42% (by HPLC). ¾ NMR (400 MHz, CDC13): δ 7.24-7.34 (m, 5H), 5.08 (m, 1H), 4.09 (m, 4H), 3.10 (m, 2H), 3.00 (s, 1H), 2.21(m, 2H), 1.82 (m, 2H), 1.46 (s, 9H), 1.23 (t, 3H). m/z: 352.20 (MH )
Example 13: Preparation of ethyl (R)-4-((2-(5-bromo)-3-(2-fluoro-6-trifluoromethyl)benzyl)-4-methyl-2,6-dioxo-3,6-dihydropyrimidin-l(2H)-yl)-l-phenylethyl)(tert-butoxycarbonyl) amino)butanoate (IV; R is ethyl)
Ethyl (R)-4-((tert-butoxycarbonyl)(2-hydroxy-l -phenyl ethyl) amino)butanoate (III; R is ethyl) (1.0 g), 5-bromo-l-(2-fluoro-6-trifluoromethyl)benzyl-6-methylpyrimidine-2,4 (1H, 3H)-dione (II) (1.08 g), Triphenyl phosphine (1.49 g) were added to THF (30 mL) at room temperature under nitrogen atmosphere. DIAD (1.11 mL) was added to the reaction mixture and stirred for 16 hours at room temperature. Water (60 volume) was added to the reaction mixture followed by ethylacetate (60 mL) was added then the layers were separated. The organic layer was dried over sodium sulfate and evaporated below 50°C under reduced pressure to obtain the crude compound. The crude compound was purified by silica gel (60/120 mesh) withl5-20% EtOAc/Hexane to obtain the title compound. Yield (1.3 g). Purity: 68.87% (by HPLC); l NMR (DMSO-d6) δ 1.15-2.0 (11H), 2.43-2.48 (4H), 3.9 (2H), 4.71-4.8 (5H), 5.3 -5.4 (3H), 7.28-7.3 (8H), 8.4 (2H); m/z: 616 (M-BOC)+
Example 14: Preparation of ethyl (R)-4-((tert-butoxycarbonyl)-2-(5-(2-fluoro-3-methoxyphenyl)-3-(2-fluoro-6-trifluoromethyl)benzyl)-4-methyl-2,6-dioxo-3,6-dihydropyrimidin-l(2H)-yl)-l-phenylethyl)amino)-butanoate (VI; R is ethyl)
Ethyl (R)-4-((2-(5-bromo)-3-(2-fluoro-6-trifluoromethyl)benzyl)-4-methyl-2,6-dioxo-3,6-dihydropyrimidin-l(2H)-yl)-l-phenylethyl)(tert-butoxycarbonyl) amino)butanoate (IV; R is ethyl) (0.9 g), 2-fluoro-3-methoxy phenyl boronic acid (V) (0.214 g) and sodium carbonate (0.797 g) were added to the mixture of 1,4-dioxane (9 mL) and water (3.06 mL) at room temperature under nitrogen atmosphere. Argon gas was bubbled through for 30 minutes. Tetrakis (triphenylphosphine)palladium (0.145 g) was added to the reaction mixture at room temperature then heated to 90-95 °C and stirred for 5 hours. The reaction mixture cooled to room temperature and filtered through celite bed then the filtrate washed with ethylacetate (9 mL) and water (36 mL) was added and stirred for 30 minutes at room temperature. Ethylacetate (36 mL) was added and the separated organic layer washed with brine and dried over sodium sulfate followed by evaporation at 45°C to obtain the crude compound. The crude compound was purified by silica gel (60/120 mesh) with 20-25% EtOAc/Hexane to obtain the title compound as yellow solid. Yield: 0.5 g; Purity: 75.1% (by HPLC); m/z: 660 (M-BOC)+.
Example 15: Preparation of ethyl (R)-4-((2-(5-(2-fluoro-3-methoxyphenyl)-3-(2-fluoro-6-trifluoromethyl)benzyl)-4-methyl-2,6-dioxo-3,6-dihydropyrimidin-l(2H)-yl)-l-phenylethyl)amino)-butanoate (VII; R is ethyl)
Ethyl(R)-4-((tert-butoxycarbonyl)-2-(5-(2-fluoro-3-methoxyphenyl)-3-(2-fluoro-6-trifluoro methyl)benzyl)-4-methyl-2,6-dioxo-3,6-dihydropyrimidin-l(2H)-yl)-l-phenylethyl)amino)-butanoate (VI; R is ethyl) (0.4 g) was added to dichloromethane (4 mL) at room temperature. The reaction mixture was cooled to 0-5 °C then trifluoroacetic acid (2 mL) was added and stirred for five hours at 0-5 °C. Saturated sodium bicarbonate solution (40 mL) was added to the reaction mixture followed by dichloromethane (40 mL) was added. The organic layer was washed with brine then dried over sodium sulfate and evaporated at 35°C to obtain the crude compound. The crude compound purified by silica gel (60/120 mesh) with 30-35% EtOAc/Hexane to obtain the title compound as yellow solid. Yield: 160 mg; Purity: 88.6% (by HPLC). ‘H NMR (400 MHz, DMSO-d6): δ 7.64 (m, 1H), 7.54 (m, 2H), 7.15-7.27 (m, 6H), 6.85 (m, 2H), 5.31 (s, 2H), 3.99 (m, 3H), 3.87 (m, 2H), 3.83 (s, 3H), 2.30-2.16 (m, 4H), 2.10 (s, 3H), 1.50 (m, 2H), 1.10 (t, 3H). m/z: 660 (MH )
PAPER
Discovery of sodium R-(+)-4-(2-(5-(2-fluoro-3-methoxyphenyl)-3-(2-fluoro-6-(trifluoromethyl-)benzyl)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-yl)-1-phenylethamino)butyrate (elagolix), a potent and orally available nonpeptide antagonist of the human gonadotropin-releasing hormone receptor
J Med Chem 2008, 51(23): 7478
Discovery of Sodium R-(+)-4-{2-[5-(2-Fluoro-3-methoxyphenyl)-3-(2-fluoro-6-[trifluoromethyl]benzyl)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-yl]-1-phenylethylamino}butyrate (Elagolix), a Potent and Orally Available Nonpeptide Antagonist of the Human Gonadotropin-Releasing Hormone Receptor
* To whom correspondence should be addressed. Phone: 1-858-617-7600. Fax: 1-858-617-7925. E-mail: cchen@neurocrine.com, sstruthers@neurocrine.com., †
Department of Medicinal Chemistry., ‡ Department of Endocrinology., § Department of Preclinical Development.
Abstract

The discovery of novel uracil phenylethylamines bearing a butyric acid as potent human gonadotropin-releasing hormone receptor (hGnRH-R) antagonists is described. A major focus of this optimization was to improve the CYP3A4 inhibition liability of these uracils while maintaining their GnRH-R potency. R-4-{2-[5-(2-Fluoro-3-methoxyphenyl)-3-(2-fluoro-6-[trifluoromethyl]benzyl)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-yl]-1-phenylethylamino}butyric acid sodium salt, 10b (elagolix), was identified as a potent and selective hGnRH-R antagonist. Oral administration of 10b suppressed luteinizing hormone in castrated macaques. These efforts led to the identification of 10b as a clinical compound for the treatment of endometriosis.
NA SALT
(R)-4-{2-[5-(2-Fluoro-3-methoxyphenyl)-3-(2-fluoro-6-[trifluoromethyl]benzyl)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-yl]-1-phenylethylamino}butyric Acid Sodium Salt
sodium salt as a white solid (1.58 g, 2.47 mmol, 62%). HPLC purity: 100% (220 and 254 nm). 1H NMR (CD3OD): 1.72 (m, 2H), 2.08 (s, 3H), 2.16 (t, J = 6.9 Hz, 2H), 2.50 (t, J = 6.9 Hz, 2H), 3.86 (s, 3H), 4.24 (m, 3H), 5.40 (d, J = 9.0 Hz, 1H), 5.46 (d, J = 9.0 Hz, 1H), 6.62 and 6.78 (m, 1H), 7.12 (m, 2H), 7.34 (m, 5H), 7.41 (m, 1H), 7.56 (m, 1H), 7.61 (d, J = 8.0 Hz, 1H). MS: 632 (M − Na + 2H+). Anal. (C32H29F5N3O5Na·0.75H2O): C, H, N, Na.
PATENT
CN 105218389
PATENT
“Elagolix” refers to 4-((R)-2-[5-(2-fluoro-3-methoxy-phenyl)-3-(2- fluoro-6 rifluoromethyl-benzyl)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-l-yl]-l- phenyl-ethylamino)-butyric acid or a pharmaceutically acceptable salt thereof. Elagolix is an orally active, non-peptide GnRH antagonist and is unlike other GnRH agonists and injectable (peptide) GnRH antagonists. Elagolix produces a dose dependent suppression of pituitary and ovarian hormones in women. Methods of making Elagolix and a pharmaceutically acceptable salt thereof are described in WO 2005/007165, the contents of which are herein incorporated by reference.
References
- ^ Jump up to:a b c d e f g Ezzati, Mohammad; Carr, Bruce R (2015). “Elagolix, a novel, orally bioavailable GnRH antagonist under investigation for the treatment of endometriosis-related pain”. Women’s Health. 11(1): 19–28. doi:10.2217/whe.14.68. ISSN 1745-5057.
- Jump up^ Chen C, Wu D, Guo Z, Xie Q, Reinhart GJ, Madan A, Wen J, Chen T, Huang CQ, Chen M, Chen Y, Tucci FC, Rowbottom M, Pontillo J, Zhu YF, Wade W, Saunders J, Bozigian H, Struthers RS (2008). “Discovery of sodium R-(+)-4-{2-[5-(2-fluoro-3-methoxyphenyl)-3-(2-fluoro-6-[trifluoromethyl]benzyl)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-yl]-1-phenylethylamino}butyrate (elagolix), a potent and orally available nonpeptide antagonist of the human gonadotropin-releasing hormone receptor”. J. Med. Chem. 51 (23): 7478–85. doi:10.1021/jm8006454. PMID 19006286.
- Jump up^ Thomas L. Lemke; David A. Williams (24 January 2012). Foye’s Principles of Medicinal Chemistry. Lippincott Williams & Wilkins. pp. 1411–. ISBN 978-1-60913-345-0.
- ^ Jump up to:a b AdisInsight: Elagolix.
- ^ Jump up to:a b c d Struthers RS, Nicholls AJ, Grundy J, Chen T, Jimenez R, Yen SS, Bozigian HP (2009). “Suppression of gonadotropins and estradiol in premenopausal women by oral administration of the nonpeptide gonadotropin-releasing hormone antagonist elagolix”. J. Clin. Endocrinol. Metab. 94 (2): 545–51. doi:10.1210/jc.2008-1695. PMC 2646513
 . PMID 19033369.
. PMID 19033369. - Jump up^ Diamond MP, Carr B, Dmowski WP, Koltun W, O’Brien C, Jiang P, Burke J, Jimenez R, Garner E, Chwalisz K (2014). “Elagolix treatment for endometriosis-associated pain: results from a phase 2, randomized, double-blind, placebo-controlled study”. Reprod Sci. 21 (3): 363–71. doi:10.1177/1933719113497292. PMID 23885105.
- Jump up^ Carr B, Dmowski WP, O’Brien C, Jiang P, Burke J, Jimenez R, Garner E, Chwalisz K (2014). “Elagolix, an oral GnRH antagonist, versus subcutaneous depot medroxyprogesterone acetate for the treatment of endometriosis: effects on bone mineral density”. Reprod Sci. 21 (11): 1341–51. doi:10.1177/1933719114549848. PMC 4212335 . PMID 25249568.
External links
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014143669A1 | Mar 14, 2014 | Sep 18, 2014 | AbbVie Inc . | Compositions for use in treating heavy menstrual bleeding and uterine fibroids |
| EP2881391A1 | Dec 5, 2013 | Jun 10, 2015 | Bayer Pharma Aktiengesellschaft | Spiroindoline carbocycle derivatives and pharmaceutical compositions thereof |
| US8084614 | Apr 4, 2008 | Dec 27, 2011 | Neurocrine Biosciences, Inc. | Gonadotropin-releasing hormone receptor antagonists and methods relating thereto |
| US8263588 | Apr 4, 2008 | Sep 11, 2012 | Neurocrine Biosciences, Inc. | Gonadotropin-releasing hormone receptor antagonists and methods relating thereto |
| US8481738 | Nov 10, 2011 | Jul 9, 2013 | Neurocrine Biosciences, Inc. | Gonadotropin-releasing hormone receptor antagonists and methods relating thereto |
| US8507536 | Aug 10, 2012 | Aug 13, 2013 | Neurocrine Biosciences, Inc. | Gonadotropin-releasing hormone receptor antagonists and methods relating thereto |
| US8952161 | Jun 5, 2013 | Feb 10, 2015 | Neurocrine Biosciences, Inc. | Gonadotropin-releasing hormone receptor antagonists and methods relating thereto |
| US9034850 | Nov 19, 2010 | May 19, 2015 | Sk Chemicals Co., Ltd. | Gonadotropin releasing hormone receptor antagonist, preparation method thereof and pharmaceutical composition comprising the same |
| US9422310 | Jan 8, 2015 | Aug 23, 2016 | Neurocrine Biosciences, Inc. | Gonadotropin-releasing hormone receptor antagonists and methods relating thereto |
 |
|
| Clinical data | |
|---|---|
| Synonyms | NBI-56418; ABT-620 |
| Routes of administration |
By mouth |
| Drug class | GnRH analogue; GnRH antagonist; antigonadotropin |
| Pharmacokinetic data | |
| Biological half-life | 2.4–6.3 hours[1] |
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C32H30F5N3O5 |
| Molar mass | 631.590 g/mol |
| 3D model (JSmol) | |
///////////////ELAGOLIX, NBI 56418, UNII:5B2546MB5Z, ABT 620, priority review status, PHASE 3, AbbVie, Neurocrine Biosciences, Endometriosis
CC1=C(C(=O)N(C(=O)N1CC2=C(C=CC=C2F)C(F)(F)F)CC(C3=CC=CC=C3)NCCCC(=O)O)C4=C(C(=CC=C4)OC)F