
















Entinostat
- BAY 86-5274
- BAY86-5274
CAS 209783-80-2
209784-80-5 (HCl)
Bayer Schering Pharma Aktiengesellschaft
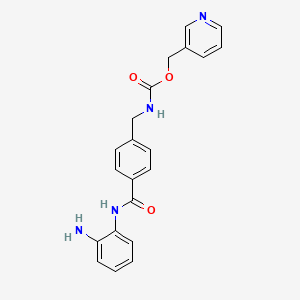
Pyridin-3-ylmethyl N-[[4-[(2-aminophenyl)carbamoyl]phenyl]methyl]carbamate
Entinostat, developed by Syndax Pharmaceuticals, is an oral selective histone deacetylase (HDAC) inhibitor primarily targeting class IHDACs (HDAC1, HDAC2, and HDAC3) . It was later licensed to
Jiangsu Hengrui Medicine Co., Ltd., for development and commercialization in China. In 2024, Entinostat has been approved by the NMPA for use in combination with exemestane to treat advanced breast cancer that is HR-positive and HER2-negative.
KHK and Syndax partner for breast cancer treatment entinostat in Japan and Korea
Japan-based Kyowa Hakko Kirin (KHK) has signed a license agreement with US-based Syndax Pharmaceuticals for the exclusive rights to develop and commercialise entinostat in Japan and Korea.
TOKYO and WALTHAM, Mass., Jan. 7, 2015 /PRNewswire/ — Kyowa Hakko Kirin Co., Ltd., (Headquarters: Chiyoda-ku, Tokyo; president and CEO: Nobuo Hanai, “Kyowa Hakko Kirin”) and Syndax Pharmaceuticals, Inc., (Waltham, Mass.; president and CEO:Arlene M. Morris, “Syndax”) today jointly announced that the companies have entered into a license agreement for the exclusive rights to develop and commercialize entinostat in Japan and Korea. Entinostat is a Class I selective histone deacetylase (HDAC) inhibitor being developed by Syndax in the United States and Europe in combination with hormone therapy for advanced breast cancer and immune therapy combinations in solid tumors.

Entinostat, also known as SNDX-275 and MS-275, is a benzamide histone deacetylase inhibitor undergoing clinical trials for treatment of various cancers.[1]
Entinostat inhibits class I HDAC1 and HDAC3 with IC50 of 0.51 μM and 1.7 μM, respectively.[2]
Entinostat (formerly known as MS-275) is a histone deacetylase (HDAC) inhibitor in phase III clincal trials at Syndax in combination with exemestane for the treatment of advanced HR-positive breast cancer.
Entinostat (MS-275) preferentially inhibits HDAC1 (IC50=300nM) over HDAC3 (IC50=8µM) and has no inhibitory activity towards HDAC8 (IC50>100µM). MS-275 induces cyclin-dependent kinase inhibitor 1A (p21/CIP1/WAF1), slowing cell growth, differentiation, and tumor development in vivo. Recent studies suggest that MS-275 may be particularly useful as an antineoplastic agent when combined with other drugs, like adriamycin.
In September 2013, Syndax Pharmaceuticals entered into a licensing, development and commercialization agreement with Eddingpharm in China and other asian countries. In 2013, a Breakthrough Therapy Designation was assigned to the compound for the treatment of locally recurrent or metastatic estrogen receptor-positive (ER+) breast cancer when added to exemestane in postmenopausal women whose disease has progressed following non-steroidal aromatase inhibitor therapy.
Clinical trials
There is an ongoing phase II trial studying the effect of entinostat on Hodgkin’s lymphoma.[3] It is in other phase II trials for advanced breast cancer (in combination with aromatase inhibitors)[4] and for metastatic lung cancer (in combination with erlotinib).[5] As of September 2013, the Food and Drug Administration is working with the industry to design phase III clinical trials. They seek to evaluate the application of Entinostat for the reduction, or prevention of, treatment resistance to aromatase inhibitors in hormone receptor positive breast cancer.[6] Syndax pharmaceuticals currently holds the rights to Entinostat and recently received $26.6 million in funds to advance treatments of resistant cancers using epigenetic tools.[7]
PHASE 3………..SYNDAX, BREAST CANCER
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Entinostat, developed by Syndax Pharmaceuticals, is an oral selec
tive histone deacetylase (HDAC) inhibitor primarily targeting class I
HDACs (HDAC1, HDAC2, and HDAC3) [7]. It was later licensed to
Jiangsu Hengrui Medicine Co., Ltd., for development and commercial
ization in China. In 2024, Entinostat has been approved by the NMPA for
use in combination with exemestane to treat advanced breast cancer that
is HR-positive and HER2-negative. This approval is specifically for pa
tients whose disease has progressed following prior endocrine therapy
[8]. Entinostat inhibits HDACs, increasing histone acetylation and
reactivating tumor suppressor genes. This mechanism restores sensi
tivity to endocrine therapy and prevents cancer cell proliferation [9].
The therapeutic agent exerts its effects by modulating the tumor
microenvironment through the suppression of immune regulatory cells,
thereby augmenting the immune response. Its clinical efficacy was
confirmed in the E2112 trial (NCT02115282), a global Phase III study.
When used in combination with exemestane, Entinostat demonstrated
the ability to extend PFS in patients with HR-positive, HER2-negative
breast cancer [10]. The median PFS was significantly extended to 6.32
months, contrasting with the 3.72 months observed in the control
cohort. In terms of safety profile, Entinostat demonstrated favorable
tolerability. The frequently encountered adverse events were primarily
neutropenia, fatigue, and nausea. Severe neutropenia occurred in 43 %
of patients but was manageable with supportive care. Liver function
abnormalities were reported but manageable with dose adjustments
[11].
The synthetic route of Entinostat is shown in Scheme 2 [12].
Enti-001 is first treated with trifluoroacetic anhydride to afford
Enti-002. Reaction of Enti-002 with oxalyl chloride yields the acyl
chloride intermediate, which undergoes condensation with Enti-003 to
form Enti-004. Subsequent alkaline hydrolysis of Enti-004 produces
Enti-005. This compound is activated with CDI followed by reaction
with Enti-006 to generate Enti-007. The synthesis concludes with acidic removal of the Boc protecting group from Enti-007, yielding Entinostat
[8] W. Li, Z. Sun, Mechanism of action for HDAC inhibitors-insights from omics
approaches, Int. J. Mol. Sci. 20 (2019) 1616.
[9] N. Bharathy, N.E. Berlow, E. Wang, J. Abraham, T.P. Settelmeyer, J.E. Hooper, M.
N. Svalina, Z. Bajwa, M.W. Goros, B.S. Hernandez, J.E. Wolff, R. Pal, A.M. Davies,
A. Ashok, D. Bushby, M. Mancini, C. Noakes, N.C. Goodwin, P. Ordentlich, J. Keck,
D.S. Hawkins, E.R. Rudzinski, A. Mansoor, T.J. Perkins, C.R. Vakoc, J.E. Michalek,
C. Keller, Preclinical rationale for entinostat in embryonal rhabdomyosarcoma,
Skelet Muscle 9 (2019) 12.
[10] B. Xu, Q. Zhang, X. Hu, Q. Li, T. Sun, W. Li, Q. Ouyang, J. Wang, Z. Tong, M. Yan,
H. Li, X. Zeng, C. Shan, X. Wang, X. Yan, J. Zhang, Y. Zhang, J. Wang, L. Zhang,
Y. Lin, J. Feng, Q. Chen, J. Huang, L. Zhang, L. Yang, Y. Tian, H. Shang, Entinostat,
a class I selective histone deacetylase inhibitor, plus exemestane for Chinese
patients with hormone receptor-positive advanced breast cancer: a multicenter,
randomized, double-blind, placebo-controlled, phase 3 trial, Acta Pharm. Sin. B 13
(2023) 2250–2258.
[11] E.T. Roussos Torres, W.J. Ho, L. Danilova, J.A. Tandurella, J. Leatherman, C. Rafie,
C. Wang, A. Brufsky, P. LoRusso, V. Chung, Y. Yuan, M. Downs, A. O’Connor, S.
M. Shin, A. Hernandez, E.L. Engle, R. Piekarz, H. Streicher, Z. Talebi, M.A. Rudek,
Q. Zhu, R.A. Anders, A. Cimino-Mathews, E.J. Fertig, E.M. Jaffee, V. Stearns, R.
M. Connolly, Entinostat, nivolumab and ipilimumab for women with advanced
HER2-negative breast cancer: a phase Ib trial, Nat Cancer 5 (2024) 866–879.
[12] T. Suzuki, T. Ando, K. Tsuchiya, T. Nakanishi, A. Saito, S. Yamashita, G. Shiraishi,
E. Tanaka, Preparation of Benzamide Derivatives as Anticancer Agents, 1998
JP10152462
SEE SCHEME AT END

Patent
http://www.google.im/patents/WO2010022988A1?cl=en
In EP 0 847 992 A1 (which co-patent is US 6,794,392) benzamide derivatives as medicament for the treatment of malignant tumors, autoimmune diseases, de- rmatological diseases and parasitism are described. In particular, these derivatives are highly effective as anticancer drugs, preferred for the haematological malignancy and solid tumors. The preparation of N-(2-aminophenyl)-4-[N- (pyridine-3-yl)methoxycarbonylaminomethyl]-benzamide is described on page 57, Example 48. The compound is neither purified by chromatography nor purified by treatment with charcoal. The final step of the process comprises the re- crystallization from ethanol.
Said compound has a melting point (mp) of 159 – 160 0C.
The IR spectrum shows the following bands: IR(KBr) cm“1: 3295, 1648, 1541 , 1508, 1457, 1309, 1183, 742.
The data indicate the Polymorph A form.
In EP 0 974 576 B1 a method for the production of monoacylated phenylenediamine derivatives is described. The preparation of N-(2- aminophenyl)-4-[N-(pyridine-3-yl)methoxycarbonylamino-methyl] benzamide is described on pages 12 to 13, Example 6. The final step of the process comprises the purification of the compound via silica gel column chromatography.
Said compound has a melting point (mp) of 159 – 160 0C.
The IR spectrum shows the following bands: IR(KBr) cm‘1: 3295, 1648, 1541 , 1508, 1457, 1309, 1183, 742.
The data indicate the Polymorph A form. In J. Med. Chem. 1999, 42, 3001-3003, the synthesis of new benzamide derivatives and the inhibition of histone deacetylase (HDAC) is described. The process for the production of N-(2-aminophenyl)-4-[N-(pyridine-3-yl) meth- oxycarbonylaminomethyl] benzamide is described. The final step of the process comprises the purification of the compound via silica gel column chromatography (ethyl acetate).
Said compound has a melting point (mp) of 159 – 160 0C.
The IR spectrum shows the following bands: IR(KBr) cm‘1: 3295, 1648, 1541 , 1508, 1457, 1309, 1183, 742.
The data indicate the Polymorph A form.
In WO 01/12193 A1 a pharmaceutical formulation comprising N-(2- aminophenyl)-4-[N-(pyridine-3-yl)methoxycarbonylamino-methyl]benzamide is described.
In WO 01/16106 a formulation comprising N-(2-aminophenyl)-4-[N-(pyridine-3- yl)methoxycarbonylamino-methyl]benzamide, having an increased solubility and an improved oral absorption for benzamide derivatives, and pharmaceutically acceptable salts thereof are described.
In WO 2004/103369 a pharmaceutical composition is described which comprises histone deacetylase inhibitors. That application concerns the combined use of N-(2-aminophenyl)-4-[N-(pyridine-3-yl)methoxycarbonylamino- methyl]benzamide together with different cancer active compounds. In fact that application is a later application, which is based on the above mentioned matter and thus concerns the Polymorph A form. Finally, JP 2001-131130 (11-317580) describes a process for the purification of monoacylphenylenediamine derivatives. In Reference Example 2, the process for the production of crude N-(2-aminophenyl)-4-[N-(pyridine-3-yl) meth-oxycarbonylaminomethyl] benzamide is described. Said compound has a melting point (mp) of 159 – 160 0C,
The IR spectrum shows the following bands: IR(KBr) cm“1: 3295, 1648, 1541 , 1508, 1457, 1309, 1183, 742.
The data indicate the Polymorph A form.
Moreover, Working Example 1 describes the purification of crude N-(2- aminophenyl)-4-[N-(pyridine-3-yl) methoxycarbonylaminomethyl] benzamide in aqueous acid medium together with carbon The final crystallization is done under aqueous conditions at 40-500C.
Following the description to that example it can be seen from the Comparative Examples 1 – 3 that the crude N-(2-aminophenyl)-4-[N-(pyridine-3-yl) meth- oxycarbonylaminomethyl] benzamide is not purified by dissolution under reflux conditions in either ethanol, methanol or acetonithle followed by a recrystalliza- tion at 2°C. As a result, these recrystallisations do not yield any pure compound.
In addition a “purification” of crude N-(2-aminophenyl)-4-[N-(pyridine-3-yl) methoxycarbonylaminomethyl] benzamide in ethanol under reflux conditions to- gether with carbon is dechbed. After filtering off the carbon the compound is re- crystallized at 2°C. The purification effect of this method is very limited. 1 ,1 % of an impurity remain in the N-(2-aminophenyl)-4-[N-(pyridine-3-yl) methoxycarbonylaminomethyl] benzamide. As a result, this procedure does not yield any pure compound.
None of the state of the art documents refer to a polymorph B of N-(2- aminophenyl)-4-[N-(pyridine-3-yl)methoxycarbonylamino-methyl]benzamide and no physicochemical features of said compound are known. Several biological and clinical studies have been done with N-(2-aminophenyl)- 4-[N-(pyridine-3-yl) meth-oxycarbonylaminomethyl] benzamide. For example, Kummar et al., Clin Cancer Res. 13 (18), 2007, pp 5411-5417 describe a phase I trial of N-(2-aminophenyl)-4-[N-(pyridine-3-yl) meth-oxycarbonylaminomethyl] benzamide in refractory solid tumors. The compound was applied orally.
The crude N-(2-aminophenyl)-4-[N-(pyridine-3-yl)methoxycarbonylaminomethyl]- benzamide of step a) can be produced according to the method described in example 6 of EP 0974 576 B1.
PATENT
http://www.google.co.in/patents/EP0974576A2?cl=en
Example 6Synthesis of N-(2-aminophenyl)-4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzamide (an example in which after activation with N,N’-carbonyldiimidazole, an acid was added to carry out reaction)
-
[0082]7.78 g (48 mmole) of N,N’-carbonyldiimidazole were added to a 1,3-dimethyl-2-imidazolidinone (50 g) suspension including 11.45 g (40 mmole) of 4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzoic acid. After stirring at room temperature for 2 hours, 17.30 g (0.16 mole) of 1,2-phenylenediamine were added to the solution. After cooling to 2°C, 9.60 g (0.1 mole) of methanesulfonic acid were added dropwise. After stirring for 2 hours, water was added, and the deposited solid was collected by filtration. Purification was then carried out through silica gel column chromatography to obtain 10.83 g (yield: 72%) of N-(2-aminophenyl)-4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzamide.
Reaction selectivity based on the result in HPLCRetention Time/min. Area % Benzoylimidazole as Active Intermediate 4.3 0.00 Monoacylated Phenylenediamine 4.7 98.91 Diacylated Phenylenediamine 11.7 1.09 Analysis data of the product
mp. 159-160°C
1H NMR (270MHz, DMSO-d6) δ ppm: 4.28 (2H, d, J=5.9Hz), 4.86 (2H, s), 5.10 (2H, s), 6.60 (1H, t, J=7.3Hz), 6.78 (1H, d, J=7Hz), 6.97 (1H, t, J=7Hz), 7.17 (1H, d, J=8Hz), 7.3-7.5 (3H, m), 7.78 (1H, d, J=8Hz), 7.93 (2H, d, J=8Hz), 8.53 (1H, d, J=3.7Hz), 8.59 (1H, s), 9.61 (1H, s).
IR (KBr) cm-1: 3295, 1648, 1541, 1508, 1457, 1309, 1183, 742
PATENT
WO 2009076206
http://www.google.com/patents/WO2009076206A1?cl=en
Suzuki et al (Suzuki et al Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives, J Med Chem 1999, 42, (15), 3001-3) discloses benzamide derivatives having histone deacetylase inhibitory activity and methods of making benzamide derivatives having histone deacetylase inhibitory activity. Suzuki et al is hereby incorporated herein by reference in its entirety.
[18] An example of the synthesis method of Suzuki et al to produce MS-275 via a three- step procedure in 50.96% overall yield is outlined in Scheme 3 below.
Scheme 3: Previous Procedure for Synthesis of MS-275 en rt, 4h
(used without purification)

[Overall yield: 0.91 x 0.56 x 100 = 50.96%;

MS-275 [19] In addition to the modest overall yield, the procedure of Suzuki et al has other disadvantages, such as a tedious method for the preparation of an acid chloride using oxalyl chloride and requiring the use of column chromatography for purification.
The synthesis of MS-275 is shown below in Scheme 4 as an example of Applicants invention of a two-step procedure: [37] Scheme 4: Preparation of MS-275
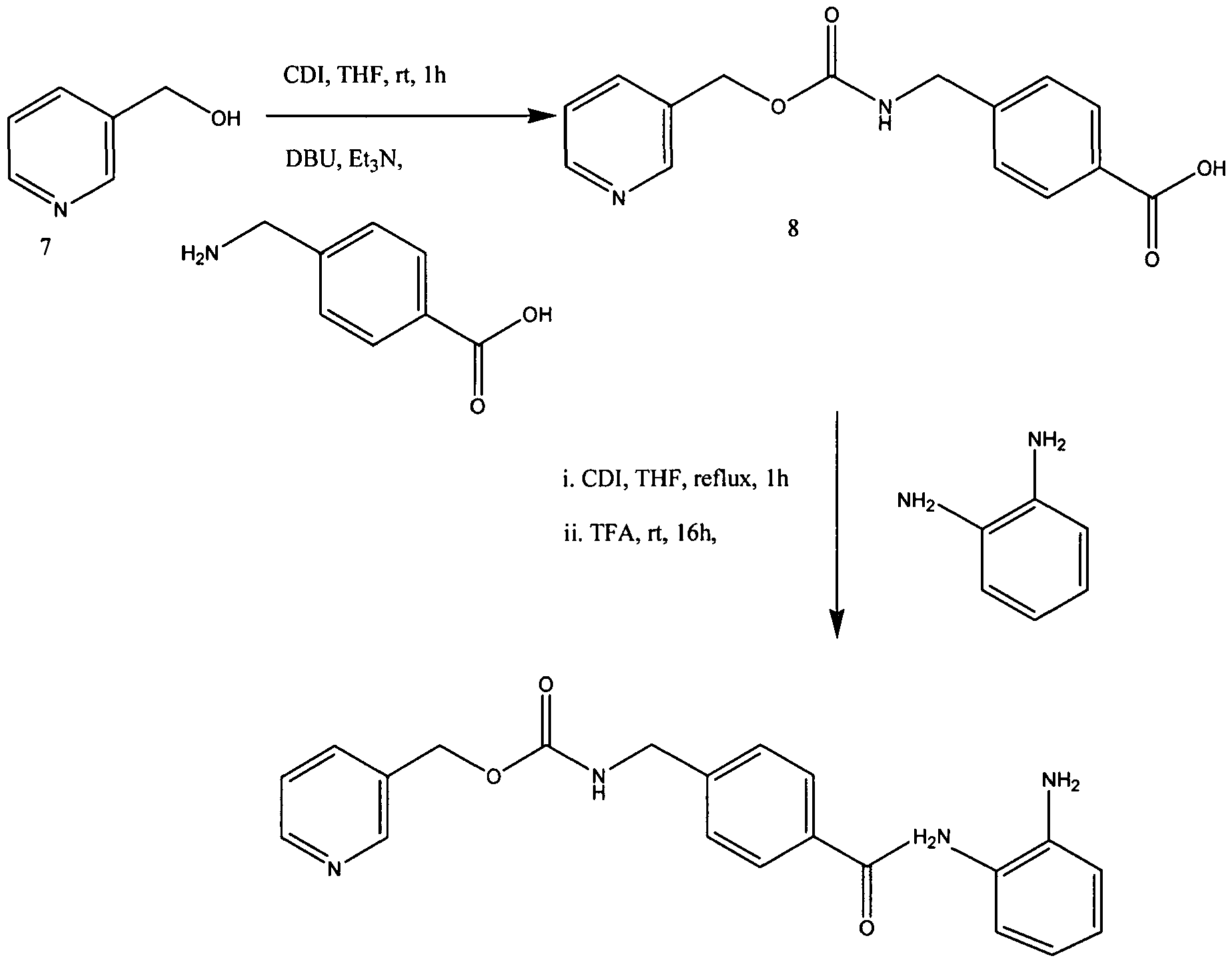
Scheme 4: New Synthesis of MS-275 (4)

Condensation of 3-(hydroxymethyl)pyridine (7) and 4-(aminomethyl)benzoic in the presence of CDI gave 4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzoic Acid (8) in 91.0% yield. In the previous method of Suzuki et ah, the carboxylic acid derivative 8 was first converted into acyl chloride hydrochloride by treatment of oxalyl chloride in toluene and then reacted with imidazole to form the acylimidazole intermediate. (Suzuki et al., Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives. J Med Chem 1999, 42, (15), 3001-3.). However, Applicants synthesized the imidazolide of intermediate 8 by treatment with CDI at about 55-60 0C in THF. The imidazolide was cooled to ambient and further reacted in situ with 1,2-phenylenediamine in the presence of TFA to afford MS-275
(4).
Experimental Section
[62] iV-(2-Aminophenyl)-4-[iV-(pyridin-3-ylmethoxycarbonyl) aminomethyl] benzamide (4, MS-275).
[63] To a suspension of 4-[N-(Pyridin-3-ylmethoxycarbonyl)aminomethyl]benzoic
Acid (5.0 g, 0.017 mol) in THF (100 mL) was added CDI (3.12 g, 0.019 mol), and the mixture stirred for 3 h at 60 0C. After formation of acylimidazole the clear solution was cooled to room temperature (rt). To this was added 1,2-phenylenediamine (15.11 g, 0.14 mmol) and trifluoroacetic acid (1.2 mL, 0.015 mol) and then stirred for 16 h. The reaction mixture was evaporated to remove THF and crude product was stirred in a mixture of hexane and water (2:5, v/v) for 1 h and filtered and dried. The residue was stirred in dichloromethane twice to afford pure MS-275 (4) as off white powder 5.25 g, 80% yield:
mp 159-160 * C; IR (KBr) 3295, 1648, 1541, 1508, 1457, 1309, 1183, 742 cm“1.
1H NMR (DMSO-J6) δ 4.28 (d, 2H, J = 5.9 Hz), 4.86 (s, 2H), 5.10 (s, 2H), 6.60 (t, IH, J = 7.3 Hz), 6.78 (d, IH, J = 7 Hz), 6.97 (t, IH, J= 7 Hz), 7.17 (d, IH, J= 8 Hz), 7.3-7.5(m, 3H), 7.78 (d, IH, J= 8 Hz), 7.93 (d, 2H, J = 8 Hz), 8.53 (d, IH, J = 3.7 Hz), 8.59 (s, IH), 9.61 (s, IH);
HRMS: calcd 376.1560 (C2iH2oN4θ3), found 376.1558. These spectral and analytical data are as previously reported in J Med Chem 1999, 42, (15), 3001-3.
[64] 4-[7V-(Pyridin-3-ylmethoxycarbonyI)aminomethyl] benzoic Acid (8) may be prepared as follows. To a suspension of l, l’-carbonyldiimidazole (CDI, 25.6 g, 158 mmol) in THF (120 mL) was added 3-pyridinemethanol (7, 17.3 g, 158 mmol) in THF (50 mL) at 10 0C, and the mixture stirred for 1 h at rt. The resulting solution was added to a suspension of 4-(aminomethyl)benzoic acid (22.6 g, 158 mmol), DBU (24.3 g, 158 mmol), and triethylamine (22.2 mL, 158 mmol) in THF (250 mL). After stirring for 5 h at rt, the mixture was evaporated to remove THF and then dissolved in water (300 mL). The solution was acidified with HCl (pH 5) to precipitate a white solid which was collected by filtration, washed with water (300 mL) and methanol (50 mL), respectively, and dried to yield pure 8 (41.1 g, 91% yield):
mp 207-208 0 C;
IR (KBr) 3043, 1718, 1568, 1434, 1266, 1 108, 1037, 984, 756 cm4; 1H NMR (DMSO-^6) δ 4.28 (d, 2H, J= 5.9 Hz), 5.10 (s, 2H), 7.3-7.5 (m, 3H), 7.7-8.1 (m, 4H), 8.5-8.7 (m, 2H). These spectral and analytical data are as previously reported in Suzuki et al, J Med Chem 1999, 42, (15), 3001-3.
PAPER
Volume 18, Issue 11, 1 June 2010, Pages 3925–3933
http://www.sciencedirect.com/science/article/pii/S0968089610003378
PAPER
see
Bioorg Med Chem 2008, 16(6): 3352
http://www.sciencedirect.com/science/article/pii/S0968089607010577
PAPER
see
Bioorganic and Medicinal Chemistry Letters, 2004 , vol. 14, 1 pg. 283 – 287
http://www.sciencedirect.com/science/article/pii/S0960894X03010539
PAPER
J Med Chem 1999, 42(15): 3001
http://pubs.acs.org/doi/abs/10.1021/jm980565u
N-(2-Aminophenyl)-4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzamide (1, MS-275). To a solution of imidazole (0.63 g, 9.2 mmol) in THF (20 mL) was added 3 (1 g, 2.9 mmol), and the mixture stirred for 1 h at room temperature. After imidazole hydrochloride was removed by filtration, 1,2-phenylenediamine (2.52 g, 23.2 mmol) and trifluoroacetic acid (0.2 mL, 2.6 mmol) were added to the filtrate and stirred for 15 h. The reaction mixture was evaporated to remove THF and partitioned between ethyl acetate (500 mL) and water (400 mL). The organic layer was washed with water and dried and then purified by silica gel column chromatography (ethyl acetate) to give 1 (0.62 g, 56% yield):
mp 159−160 °C;
1H NMR (DMSO-d6) δ 4.28 (d, 2H, J = 5.9 Hz), 4.86 (s, 2H), 5.10 (s, 2H), 6.60 (t, 1H, J = 7.3 Hz), 6.78 (d, 1H, J = 7 Hz), 6.97 (t, 1H, J = 7 Hz), 7.17 (d, 1H, J = 8 Hz), 7.3−7.5(m, 3H), 7.78 (d, 1H, J = 8 Hz), 7.93 (d, 2H, J = 8 Hz), 8.53 (d, 1H, J = 3.7 Hz), 8.59 (s, 1H), 9.61 (s, 1H);
IR (KBr) 3295, 1648, 1541, 1508, 1457, 1309, 1183, 742 cm-1.
Anal. (C21H20N4O3) C, H, N.
………………………………………………………………………..
see
Bulletin of the Korean Chemical Society, 2014 , vol. 35, 1 pg. 129 – 134
http://koreascience.or.kr/article/ArticleFullRecord.jsp?cn=JCGMCS_2014_v35n1_129
PAPER
see
ChemMedChem, 2013 , vol. 8, 5 pg. 800 – 811
PAPER
see
ACS Medicinal Chemistry Letters, 2013 , vol. 4, 10 pg. 994 – 999
http://pubs.acs.org/doi/full/10.1021/ml400289e
References
- Phase I trial of 5-azacitidine (5AC) and SNDX-275 in advanced lung cancer (NSCLC)
- Novel Sulphonylpyrroles as Inhibitors of Hdac S Novel Sulphonylpyrroles
- A Phase 2 Multi-Center Study of Entinostat (SNDX-275) in Patient With Relapsed or Refractory Hodgkin’s Lymphoma
- A Phase 2, Multicenter Study of the Effect of the Addition of SNDX-275 to Continued Aromatase Inhibitor (AI) Therapy in Postmenopausal Women With ER+ Breast Cancer Whose Disease is Progressing
- A Phase 2 Exploratory Study of Erlotinib and SNDX-275 in Patients With Non-small Cell Lung Carcinoma Who Are Progressing on Erlotinib
- Breakthrough Designation Granted to Entinostat for Advanced Breast Cancer Silas Inman Published Online: Wednesday, September 11, 2013 http://www.onclive.com/web-exclusives/Breakthrough-Designation-Granted-to-Entinostat-for-Advanced-Breast-Cancer
- http://www.syndax.com/assets/130827%20Syndax%20Series%20B%20news%20release.pdf
- References:
1. Saito, A. et al. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc Natl Acad Sci USA 96 4592-4597 (1999).
2. Jaboin, J., et al. MS-27-275, an inhibitor of histone deacetylase, has marked in vitro and in vivo antitumor activity against pediatric solid tumors. Cancer Res 62 6108-6115 (2002).
3. Rosato RR, et al. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res 2003; 63: 3637–3645.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| EP0847992B1 * | Sep 30, 1997 | Jun 23, 2004 | Schering Aktiengesellschaft | Benzamide derivatives, useful as cell differentiation inducers |
| US7244751 * | Feb 2, 2004 | Jul 17, 2007 | Shenzhen Chipscreen Biosciences Ltd. | N-(2-amino-5-fluorophenyl)-4-[N-(Pyridn-3-ylacryloyl)aminomethyl]benzamide or other derivatives for treating cancer and psoriasis |
| Reference | ||
|---|---|---|
| 1 | * | MAI A: ‘Histone deacetylation in epigenetics: an attractive target for anticancer therapy‘ MED RES REV. vol. 25, no. 3, May 2005, pages 261 – 309 |
| 2 | * | SUZUKI T ET AL.: ‘Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives‘ J MED CHEM. vol. 42, no. 15, 29 July 1999, pages 3001 – 3003 |

| Names | |
|---|---|
| Preferred IUPAC name(Pyridin-3-yl)methyl ({4-[(2-aminophenyl)carbamoyl]phenyl}methyl)carbamate | |
| Other namesSNDX-275; MS-275 | |
| Identifiers | |
| CAS Number | 209783-80-2 |
| 3D model (JSmol) | Interactive image |
| ChEBI | CHEBI:132082 |
| ChEMBL | ChEMBL27759 |
| ChemSpider | 4111 |
| ECHA InfoCard | 100.158.999 |
| IUPHAR/BPS | 7007 |
| KEGG | D09338 |
| PubChem CID | 4261 |
| UNII | 1ZNY4FKK9H |
| CompTox Dashboard (EPA) | DTXSID0041068 |
| InChI | |
| SMILES | |
| Properties | |
| Chemical formula | C21H20N4O3 |
| Molar mass | 376.4085 g/mol |
| Pharmacology | |
| ATC code | L01XH05 (WHO) |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
{kind=link}
/////////////////approvals 2024, china 2024, Entinostat, SNDX 275, MS 275, Syndax, BAY 86-5274, BAY86-5274



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……