














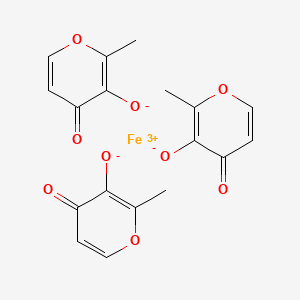
Ferric Maltol
Iron, tris(3-hydroxy-2-methyl-4H-pyran-4-onato-O3,O4)-
| Molecular Formula: | C18H15FeO9 |
|---|---|
| Molecular Weight: | 431.154 g/mol |
iron(3+);2-methyl-4-oxopyran-3-olate
RN: 33725-54-1
UNII: MA10QYF1Z0
Feraccru
Ferric maltol; UNII-MA10QYF1Z0; MA10QYF1Z0; Ferric maltol (INN); Ferric maltol [INN]; 33725-54-1
Shield Therapeutics, under license from Vitra Pharmaceuticals
UPDATE
- Originator University of Cambridge; University of London
- Developer Shield Therapeutics
- Class Antianaemics; Ferric compounds; Pyrones; Small molecules
- Mechanism of Action Iron replacements
- Marketed Iron deficiency anaemia
Most Recent Events
- 26 Jul 2019 Registered for Iron deficiency anaemia (In adults) in USA (PO)
- 24 Apr 2019 Swissmedic approves a extension of the approved indication for ferric maltol to include treatment of all adults with iron deficiency (ID) with or without anaemia
- 14 Mar 2019 The European Patent Office decides in favour of Shield Therapeutics in relation to the group’s patent No. 2 668 175
The US Food and Drug Administration (FDA) has approved oral ferric maltol(Accrufer, Shield Therapeutics) AUG 2019 for the treatment of iron deficiency in adults.
The product is already approved in the European Union and Switzerland for the treatment of iron deficiency in adults, where it is sold as Feraccru.
OLD DATA NDA filing expected in US in 2H 2018, Ph 3 trial is planned in 2018/19 for treatment of iron deficiency anemia (IDA) in children., Expected dose form: Oral Capsule; 30 mg
Treatment of iron deficiency anemia (IDA) associated with inflammatory bowel disease (IBD) and Chronic Kidney disease.
Iron deficiency anaemia (IDA) occurs when iron levels are insufficient to support red blood cell production and is defined – according to the WHO – as haemoglobin levels below 13 g/dL in men over 15 years, below 12 g/dL in non-pregnant women over 15 years, and below 11 g/dL in pregnant women. Iron is absorbed at the apical surface of enterocytes to be transported by ferroportin, the only known iron exporter, across the basolateral surface of the enterocyte into circulation. Inflammation from IBD interferes with iron absorption by causing an increase in hepcidin, a peptide hormone synthesized in the liver that inhibits ferroportin activity. Anaemia is the most common extra-intestinal complication of inflammatory bowel disease (IBD) and although it often involves a combination of IDA and anaemia of chronic disease, IDA remains an important contributor in this condition due to chronic intestinal bleeding and decreased iron intake (from avoidance of foods that may exacerbate symptoms of IBD). In a variety of populations with IBD, the prevalence of iron deficiency anaemia ranges from 36%-76%. The serum markers of iron deficiency are low ferritin, low iron, raised total iron binding capacity, raised red cell protoporhyrin and increased transferrin binding receptor (sTfR). Serum ferritin is the most powerful test for iron deficiency. The cut-off level of ferritin which is diagnostic varies between 12-15 µg/L. Higher levels of serum ferritin do not exclude the possibility of iron deficiency, and a serum ferritin level of <100 μg/L may still be consistent with iron deficiency in patients with IBD. A transferrin saturation of <16% is indicative of iron deficiency, either absolute or functional. Other findings on a complete blood count panel that are suggestive of iron deficiency anaemia, but are not considered diagnostic, include microcytosis, hypochromia, and elevation of red cell distribution width.
A deficiency of iron can have a significant impact on a patient’s quality of life. Appropriate diagnosis and treatment of iron deficiency anaemia are important to improve or maintain the quality of life of patients. The goals of treatment are to treat the underlying cause, limit further blood loss or malabsorption, avoid blood transfusions in haemodynamically stable patients, relieve symptoms, and improve quality of life. More specifically, therapeutic goals of treatment include normalizing haemoglobin levels within 4 weeks (or achieving an increase of >2 g/dL) and replenishing iron stores (transferrin saturation >30%). Oral iron supplementation has been considered standard treatment because of an established safety profile, lower cost, and ease of administration. It has been shown to be effective in correcting anaemia and repleting iron stores. One concern with higher doses of daily oral iron is intolerance due to GI side effects. Symptoms include nausea, vomiting, diarrhea, abdominal pain, constipation, and melena-like stools. Guidelines on the Diagnosis and Management of Iron Deficiency and Anaemia in Inflammatory Bowel Diseases recommend IV iron therapy over oral iron supplementation in the treatment of iron deficiency anaemia in patients with IBD, citing faster and prolonged response to treatment, decreased irritation of existing GI inflammation, improved patient tolerance, and improved quality of life. Patients with severe anaemia (haemoglobin level of <10 g/dL), failure to respond or intolerance to oral iron therapy, severe intestinal disease or patients receiving concomitant erythropoietin are recommended indications for IV iron therapy. Other conditions where patients should be considered for first-line IV therapy over oral therapy include congestive heart failure, upper GI bleeding, and in situations where rapid correction of anaemia may be required.
Across EU there are several iron (Fe+2) oral preparations as ferrous fumarate, ferrous gluconate, ferrous sulphate and ferrous glycine sulfate, formulated as tablet, solution or gastroresistent capsules. All ferrous compounds are oxidised in the lumen of the gut or within the mucosa with release of activated hydroxyl radicals, which may attack the gut wall and can effect a range of gastrointestinal symptoms and discomfort. Ferric preparations also exist but with less bioavailability. Across EU there are also several IV products on the market: iron (III) hydroxide dextran complex, iron sucrose, ferric carboxymaltose, iron isomaltoside. IV iron therapy, however, is inconvenient, invasive and associated with the risk of rare but serious hypersensitivityreactions; it is used in those situations when oral preparations cannot be used or when there is a need to deliver iron rapidly. Feraccru is a trivalent iron, oral iron replacement preparation. The active substance of Feraccru is ferric maltol (also known as 3-hydroxy-2-methyl-4H-pyrane-4-one iron (III) complex, or ST10, or ferric trimaltol or ferric maltol) an oral ferric iron/maltol complex. It is presented as red hard gelatine capsules containing 30 mg iron (ferric iron). Maltol is a sugar derivative that strongly chelates iron in the ferric form (FeIII) rendering the iron stable and available for absorption. Upon dissociation of the ferric maltol complex, the maltol molecules are absorbed and glucuronidated in the intestinal wall, and within the liver during first pass metabolism, and subsequently eliminated from the body in the urine. The iron is absorbed via the endogenous dietary iron uptake system. The indication finally agreed with the CHMP was: Feraccru is indicated in adults for the treatment of iron deficiency anaemia (IDA) in patients with inflammatory bowel disease (IBD) (see section 5.1). The proposed dosage is one 30 mg capsule twice daily on an empty stomach, corresponding to 60 mg ferric iron per day. There was agreement in the paediatric investigation plan to grant a deferral and a waiver for iron as iron (III)-maltol complex (EMEA-001195-PIP01-11).The PDCO granted a waiver in infants under 6 months of age and a referral for the completion of the planned paediatric studies (ST10-021 PK-PED/ST10-01-102, an open label, randomised, multiple-dose, parallel PK study and ST10-01-303, a randomised, open label comparative safety and efficacy study of ST10 and oral ferrous sulphate as comparator) until the adult studies are completed.
SYN
Patent
https://patents.google.com/patent/WO2017167963A1/en
The sugar derivative maltol is a hydroxypyrone (IUPAC name: 3- hydroxy-2-methyl-4£f-pyran-4-one) and it strongly chelates iron and the resulting complex (ferric trimaltol) is well absorbed, unlike many other ferric iron therapies. Ferric trimaltol appears well tolerated even in populations highly susceptible to gastrointestinal side-effects, such as IBD patients (Harvey et al . , 1998), and as such it provides a valuable alternative to patients who are intolerant of oral ferrous iron products, notably in place of intravenous iron. Clinical trials using ferric trimaltol have been carried out, see for example, Gasche et al., 2015.
However, despite the evidence of bioavailability and tolerability for ferric trimaltol, its clinical development has been limited by the absence of adequate synthetic routes. In particular, most manufacturing processes require the use of organic solvents, which increase manufacturing costs, for example to deal with post-synthesis solvent removal, and require additional safety measures, for example to deal with flammability . Critically, solvent-based syntheses are not robust and often generate ferric hydroxide, described in the prior art to be an unwanted impurity of the synthesis.
WO 03/097627 (Vitra Pharmaceuticals Limited) describes the synthesis of ferric trimaltol from iron salts of carboxylic acids in aqueous solution at a pH greater than 7. In a first
synthesis, ferric citrate is added to a solution of sodium hydroxide at room temperature and maltol is added to a second solution of sodium hydroxide at pH 11.6. The ferric citrate solution is added to the maltol solution, leading to the
production of a deep red precipitate. This composition is then evaporated until dryness and the material is powdered and dried. Alternative syntheses are described using ferrous fumarate or ferrous gluconate as the iron carboxylate salt starting material, and by dissolving maltol in sodium carbonate solution in place of sodium hydroxide. However, despite the fact that this process is fully aqueous, several of the iron carboxylate salts employed are expensive, especially as they need to be pharmaceutical grade if the ferric trimaltol is to be suitable for human administration. More importantly, this process introduces high levels of
carboxylates (equimolar to iron or greater) to the synthesis that are not easily removed by filtration or centrifugation of the ferric trimaltol cake. Instead these water soluble contaminants must be washed off (e.g. water washed), but this would result in considerable losses of the product due to the amphipathic nature of ferric trimaltol.
WO 2012/101442 (Iron Therapeutic Holdings AG) describes the synthesis of ferric trimaltol by reacting maltol and a non- carboxylate iron salt in an aqueous solution at alkaline pH .
However, despite the lower cost of non-carboxylate iron salts, pharmaceutically appropriate grades are still required if the ferric trimaltol is to be suitable for human administration and hence are comparatively expensive starting materials.
Importantly, the use of non-carboxylate iron salts (e.g. ferric chloride) results in the addition of considerable levels of the respective counter-anion (e.g. three moles of chloride per every mole of iron) of which a significant part is retained in the filtration (or centrifugation) cake and thus must be washed off. As such, WO 2012/101442 does not address the problem of product losses in WO 03/097627. Furthermore, the addition of a non- carboxylate iron salt (e.g. ferric chloride) to a very alkaline solution, as described in WO 2012/101442, promotes the formation of stable iron oxides, which is an unwanted contaminant in ferric trimaltol . As a consequence, further costly and time-consuming processing of the material would be required for manufacturing .
Overall, the cost of the current aqueous syntheses is driven by regulatory demands for low levels of toxic heavy metals and residual reagents in the final pharmaceutical formulation, which force the use of highly purified, and thus expensive, iron salts as well as thorough washing of the final product (resulting in significant losses of product) . This will impact on the final price of ferric trimaltol and potentially limits patient access to this therapy. As such, there is a need for a process that can use lower iron grades and limited wash cycles, whilst producing ferric trimaltol of adequate purity.
Ferric maltols are a class of compounds that include ferric trimaltol, a chemical complex formed between ferric iron (Fe3+) and the hydroxypyrone, maltol (IUPAC name: 3-Hydroxy-2-methyl-4£f- pyran-4-one) , in a molar ratio of ferric iron to maltol of 3:1. Maltol strongly chelates the ferric iron and the resulting complex (ferric trimaltol which may also be written as ferric tri-maltol) is well absorbed, in contrast to some other ferric iron supplements, fortificants and therapies. Maltol binds metal cations mainly in the form of a dioxobidentate ligand in a similar manner proposed for other 4 ( 1H) -pyranones :

Structure of maltol (3-hydroxy-2-methyl-4 (H) -pyran-4-one) and dioxo-chelation to metal cations (M) such as iron. For ferric trimaltol three maltol groups surround one iron.
Examples
Example 1: Ferric trimaltol from L-lyslne coated ferric hydroxide
Synthesis of lysine-coated ferric hydroxide colloid
14.87g FeCl3. 6H20 was added to 25 mL UHP water and stirred until dissolved. 14.9g NaOH 5M was then added drop-wise to this solution with constant stirring, during which a ferric hydroxide colloid was gradually produced. This colloidal suspension was then added to a L-Lysine suspension (5.02g in 25mL ddH2<D) .
Ferric trimaltol synthesis
7 g NaOH pellets was added to 25 mL UHP water and stirred until dissolved. Next, 24.5g maltol was added and stirred until dissolved. Then, the suspension of lysine-coated ferric
hydroxide colloids was gradually added to the maltol with vigorous stirring, producing a dark red precipitate (with a significant brown hue) . This suspension was incubated overnight during which time it became lighter and the brown hue
disappeared. This precipitate was then recovered by
centrifugation (4500 rpm x 5min) and dried overnight (50°C) .
Example 2: Ferric trimaltol from L-lysine modified ferric hydroxide
Synthesis of lysine-modified ferric hydroxide gel
14.87g FeCl3.6H20 and 5.02g L-Lysine were added to 25 mL UHP water and stirred until dissolved. 32 mL NaOH 5M was then gradually added to this solution producing a ferric hydroxide gel .
Ferric trimaltol synthesis
7 g NaOH pellets was added to 25 mL UHP water and stirred until dissolved. Next, 24.5g maltol was added and stirred until dissolved. Next, the lysine-modified ferric hydroxide gel was gradually added to this solution with vigorous stirring. A 1.2 M HC1 solution was then used to drop the pH of the solution to 10, which was then incubated for 70 min. Finally, a dark red precipitate (i.e., ferric trimaltol) was recovered by
centrifugation (4500 rpmx5min) and dried overnight (45°C) .
Example 3: Absence of ferric hydroxide in ferric trimaltol
Ferric trimaltol is soluble in ethanol whereas ferric hydroxide (a potential contaminant) is not. As such ferric trimaltol powders produced as per Examples 1 and 2 were dissolved in ethanol. The material from Example 2 dissolved completely confirming the absence of iron hydroxides whereas the material from Example 1 did not. This supported the preference in the present invention for ligand modification, rather just surface coating, to ensure full conversion to ferric trimaltol .
Example 4: Ferric trimaltol from tartrate-modified ferric hydroxide
Synthesis of tartrate-modified ferric hydroxide gel
14.87g FeCl3.6H20 (0.055 mol) was added to 25 mL UHP water and stirred until dissolved. 4.12 g tartaric acid (0.0275 mol) was added to this solution and stirred until dissolved. 38 mL NaOH 5M was then gradually added to this solution producing a ferric hydroxide gel .
Ferric trimaltol synthesis
2 g NaOH pellets was added to 25 mL UHP water and stirred until dissolved. Next, 24.5g maltol was added and stirred. This produced a slurry in which most of the maltol remained
undissolved. Next, the tartrate-modified ferric hydroxide gel was gradually added to this solution with vigorous stirring during which the remainder of maltol dissolved. After 15 min a dark red precipitate (i.e. ferric trimaltol) had been formed and pH had stabilised at 8.5. The material was then washed by (1)
centrifuging, (2) disposing of the supernatant and (3)
resuspending in water back to its original volume. Finally, the material was recovered by centrifugation (4500 rpm x 5min) and dried overnight (50°C) . Previously disclosed synthetic processes for the production of ferric trimaltol under aqueous conditions require the addition of NaOH (or other suitable bases) for conversion of maltol from its protonated form to its deprotonated form prior to complexation of iron. However this results in the formation of unwanted sodium ions which must be washed off. In contrast, the use of ferric hydroxides according to the methods of the present invention reduces the requirements for base and associated counter cation (e.g. sodium), which is a favourable feature. Note that ferric hydroxides are represented above as Fe (OH) 3 for illustrative purposes only. Different iron hydroxides possess different structures and elemental compositions (see Cornell & Schwertmann, The Iron Oxides Structure, Properties, Reactions, Occurrence and Uses. 2nd edition, 1996, VCH Publishers, New York) . Example 5: Ferric trimaltol from tartrate-modified ferric hydroxide (with removal of contaminants from ferric hydroxide)
Material prepared as in Example 4, except excess reactants and reaction products (e.g. unbound tartaric acid, sodium chloride) were removed from the ferric hydroxide gel. This was achieved by centrifuging the ferric hydroxide gel after its synthesis and discarding the supernatant, which contained unwanted soluble species. Finally, the ferric hydroxide gel was re-suspended in water back to its original volume prior to being added to a maltol slurry.
Example 6: Ethanolic clean up for ferric trimaltol produced from ligand coated ferric hydroxide
Ferric trimaltol precipitate was purified as it contained an unwanted iron oxide fraction. Part of the wet pellet recovered by centrifugation (4.5 g) was dissolved in 1L ethanol. The iron oxide fraction (which remained undissolved) was then removed by filtration, producing a turbidity-free solution. Next, ethanol was evaporated (40°C in a rotavapor under vacuum) producing a concentrated ferric trimaltol slurry. This was then recovered and oven dried overnight at 50°C.
PATENT
https://patents.google.com/patent/WO2012101442A1/en
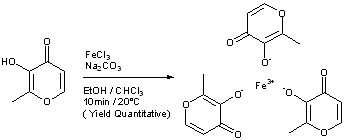
Comparative Example 1
Preparation of Iron Trimaltol from Pure Maltol Maltol was dissolved in an aqueous solution of ferric chloride and ferric trimaltol was precipitated upon the addition of sodium hydroxide.
An accurate mass of ferric chloride hexahydrate granules (330g) was dissolved in distilled water to yield a pH of 0.6. To this solution, an equimolar amount of maltol was added (490g in total, initially 250g) and allowed to dissolve with continuous stirring. The pH of this solution was found to be zero and the colour of this solution was deep- purple. Spectroscopy showed that the initial solution was mainly a 1 :1 Fe/maltol mixture with some 1 :2 component. The remaining maltol was added. After an hour of stirring, sodium hydroxide (147g NaOH in 750 ml water) was added dropwise to the solution until a pH of 8.3 was achieved. The solution and precipitate were red. The precipitate was collected using a Buchner funnel under vacuum. The precipitate was dried at 40°C under vacuum.
Maltol is only slightly soluble in an aqueous acidic reaction medium. After an hour of stirring, traces of undissolved maltol were visible on the surface of the ferric chloride/maltol solution, on the walls of the reaction vessel and on the stirrer. Upon addition of sodium hydroxide, there appeared to be lumps of a brownish-black substance on the walls of the reaction vessel and on the stirrer which seemed to add to the impurities in the desired product.
An attempt to heat the ferric chloride/maltol solution so as to assist the maltol to dissolve in the ferric chloride solution resulted in a burnt, off spec, colour iron maltol sample. This method also produces two by-products which consume expensive maltol namely Fe(OH)2 (Maltol) and Fe (OH) (Maltol)2.
The sodium hydroxide solution has to be added extremely slowly to prevent “gumming up” and formation of undesirable lumps at the bottom of the reaction vessel. A yield of about 78% ferric trimaltol was obtained using this method of preparation.
When maltol is added to a ferric chloride solution at a low pH, no ferric trimaltol is formed and ferric hydroxide is generated with ferric monomaltol and a small percentage of ferric dimaltol species. The charge neutralisation of these complexes is either the hydroxy! functional group or the chloride anion. This addition also results in the formation of black deposits and gums consisting of ferric chloride/ferric hydroxide polymers. These black deposits are also produced if the solutions are heated. Therefore it is not possible to obtain the correct stoichiometry for the formation of ferric trimaltol and manufacture a pharmaceutically acceptable product using this method.
The addition of maltol to an aqueous solution of ferric or ferrous chloride was deemed impractical for scale up and manufacturing purposes and Examples 2 to 4 investigate the addition of the iron chlorides to maltol in solution.
The problem of working in an aqueous environment
Ferric chloride as a hydrated ion in aqueous solution is a strong Lewis acid with a Ka of 7x 103 and ferrous chloride as a hydrated ion in aqueous solution is also a strong Lewis acid with a Ka of 5 x 10“9. Over the desired range for using iron chlorides as starting materials for the synthesis of ferric trimaltol, ferric chloride in aqueous solution has a pH value in the range of 1-3 and ferrous chloride has a pH in the range of 3-5. Furthermore, commercial solutions of iron chlorides have a pH circa 1 because they are stabilised by the addition of hydrochloric acid to prevent the precipitation of ferric hydroxide species.
The present invention recognises that maltol is virtually insoluble at these low pH values and has limited solubility when dissolved in water in the pH range 6-8. The maximum aqueous solubility is 1g/100m! at 20°C. However, the solubility of maltol can be increased to 10g/100ml by heating to near boiling temperatures. Maltol is stable in aqueous solution at these temperatures and this property has been employed in Example 4 to synthesise ferric trimaltol. At low pH values ferric trimaltol is not the preferred species due to disproportionation. In order to obtain significant amounts of ferric trimaltol using a stoichiometric ratio of iron salt to hydroxypyrone of 1 :3, the eventual pH of the solution must exceed 7 since below that pH ferric dimaltol and monomaltol species will exist. Therefore two methods of increasing the pH were researched 1) using sodium carbonate and 2) using sodium hydroxide. Other alkali hydroxides could be used such as potassium hydroxide. The sodium carbonate neutralisation was found to be less preferable due to C02 generation. This research lead to an improved synthesis of ferric trimaltol.
Example 2
Maltol was dissolved in an aqueous solution of sodium hydroxide and iron maltol was precipitated upon the addition of ferric chloride.
In view of some of the difficulties experienced in Example 1 , and the fact that maltol is very soluble in aqueous alkali hydroxide solutions, it was decided to change the manufacturing procedure.
The initial work using this method of preparation showed that a 90% yield was achieved. Various operating parameters were then optimised and the following procedure outlines the final method chosen. A yield of 95% was then achieved. An accurate mass of sodium hydroxide pellets (20g) was dissolved in distilled water to yield a pH of 13.50. An equimolar amount of maltol (63g) was added to this aqueous solution of NaOH to give a clear yellow coloured solution with a pH of 11.6. Almost immediately a stoichiometric amount of ferric chloride (45g) was added slowly to this solution to give a pH of 7.1 and a red precipitate formed, which was then collected using a Buchner funnel under vacuum. The precipitate was then dried at 40°C under vacuum.
Adding the maltol solution in sodium hydroxide to ferric chloride as in method 1 is not preferred since it gives an off spec product and gums and a black precipitate.
Maltol is very soluble in aqueous alkali hydroxide solutions giving a yellow solution. The concentration of the hydroxide solution preferably does not exceed 20%.
This method is advantageous since it has the potential to produce only one by-product viz, ferric hydroxide Fe(OH)3 which consumes some of the iron intended to complex with the maltol. This is not easily measurable in the presence of iron maltol and so the following method was used to measure the ferric hydroxide. Fe(OH)3 is insoluble in ethanol and so the iron maltol product was dissolved in ethanol. It was found that small amounts of Fe(OH)3 may be present in the batches of iron maltol synthesized according to Example 2.
Taking the extremes of the specification, in one embodiment, the amount of Fe(OH)3 present in the active material may not exceed 2 wt. % Fe(OH)3 based on the total weight of the composition. In view of its well known inert characteristics the level of this compound is adequately controlled and a final specification including controlled ferric hydroxide should be acceptable.
The mass balance for maltol and iron was closed at 99%.
A yield of 95% iron maltol was obtained using this method of preparation.
Example 3
Maltol was dissolved in an aqueous solution of Sodium Carbonate and Iron Maltol was precipitated upon the addition of Ferric Chloride.
An accurate mass of sodium carbonate (Na2C03) (53g) was dissolved in distilled water to give a solution having pH = 11.5. An equimolar amount of maltol (65g) was added to this aqueous alkali solution to give a murky creme coloured solution of pH = 9.9. A stoichiometric amount of a ferric chloride solution was added drop wise to this solution to a pH of 8.00. A further 15 grams of Na2C03 was added to this solution to increase the pH to 9.00. The remainder of the ferric chloride solution was then added to give a solution pH = 8.77 and a red coloured precipitate appeared.
The precipitate was collected using a Buchner funnel under vacuum. The precipitate was then dried at 40°C under vacuum. The release of C02 during the reaction tends to make this process less desirable due to foaming on the surface. The final product is a gellike solid when wet and the removal of moisture during drying can therefore be time consuming. The process may not be preferred but the ferric trimaltol produced could be acceptable.
Example 4 Maltol was dissolved in water and heated to a near boiling temperature and ferric or ferrous chloride was added to form a 1 :1/1:2 mixture of ferric maltol. The solution was allowed to cool and was added to maltol dissolved in sodium hydroxide. Stage 1
Depending on the batch size required, the ferric chloride was added slowly to a maltol solution in water at a pH of 6-7. The solubility of maltol is greatly enhanced up to 10g/100ml by heating to temperatures above 60°C. Addition of ferric chloride or ferrous chloride and monitoring the pH of the solution and maintaining the pH> 3 mainly produces ferric dimaltol species but very little ferric trimaltol. Above pH 3, no ferric hydroxide appeared to be generated. Ferric monomaltol and dimaltol species either with hydroxy or chloride giving the charge neutralisation are very soluble and a concentrated solution in excess of 30g/100ml can be generated. In order to obtain the correct stoichiometry for the formation of ferric trimaltol, further maltol is required and the pH needs to be corrected to values higher than 7.
As anhydrous ferrous or ferric chloride either 126g or 162g in 200ml of water can be added to a litre of water containing 120g of maltol. This ratio of iron to maltol does not provide sufficient maltol to produce any significant amounts of ferric trimaltol which does not precipitate at this stage.
Stage 2 Maltol in alkaline solution has been described as set out above. Conveniently, because maltol solutions up to 20% in sodium hydroxide have a pH circa 11.6, mixing of this solution with the ferric mono/dimaltol solutions from stage 1 yields a precipitate of ferric trimaltol with a deep characteristic burgundy red colour of high purity as determined by UV-vis spectroscopy. The filtrate yields product which is suitable for a GMP (good manufacturing process). The sodium chloride which is generated by this process is found in the supernatant since it has a much higher solubility at 35g/100ml than ferric trimaltol. The small amounts of sodium chloride in the ferric trimaltol can be reduced, if required, by washing in water. A further, surprising feature of the research resulted from work on ferrous chloride. Ferrous chloride may be substituted in stage 1 to form ferric dimaltol since the maltol was found to auto-oxidise the ferrous to ferric during the process of chelation. One aspect of this work which was considered to be potentially very useful if larger batch sizes were required arose from the finding that being a weaker Lewis acid than ferric chloride the pH of the starting solution was in excess of 3. Therefore the risk of generating ferric hydroxide was lower than with the use of ferric chloride at higher concentrations.
Ferrous and ferric chloride in solution or as a solid may be added to an alkaline solution of maltol in sodium hydroxide, combining stages 1 & 2. Providing a small excess of maltol up to about 10% is added then a precipitate of ferric trimaltol with a small amount of maltol is obtained. Such a preparation would be satisfactory as a GMP ferric trimaltol product.
PATENT
https://patents.google.com/patent/WO2017167963A1/en
AMPLE 1
Synthesis of ferric trimaltol using ferric citrate
NaOH (12g, 0.3 moles) is dissolved in water (50 ml) to form a sodium hydroxide solution. 20 ml of the sodium hydroxide solution is placed in a separate vessel.
Ferric citrate (30g, 0.11 moles) is slowly added to the sodium hydroxide solution in the separate vessel at room temperature with gentle stirring. Further portions of the sodium hydroxide solution are added to the solution of ferric citrate, as necessary, in order to ensure that all of the ferric citrate is dissolved.
Maltol (49g, 0.39 moles) is added to the remaining volume of sodium hydroxide solution and dissolved. The pH of the maltol solution is 11.6.
The ferric citrate solution is slowly added to the maltol solution with gentle stirring. A deep red precipitate forms; the supernatant is a deep red colour.
The solution is slowly evaporated to dryness at 60 to 80° C until the material is suitable for powdering. The material is powdered and the powder is then dried to a constant weight.
The yield of the final product is 87g. The final product comprises ferric trimaltol and sodium citrate. The product was assayed, using elemental analysis, for iron and sodium content. The iron content is 7.89% (theoretical 7.8%) and the sodium content is 13.45%.
The pH of a solution of the final product in water was measured. The pH of a 1% solution of the product by total weight of aqueous solution is 9.9 at 20°C.
EXAMPLE 2
Synthesis of ferric trimaltol using ferrous fumarate
NaOH (40g, 1 mole) is dissolved in water (100 ml) to form a sodium hydroxide solution. The pH of the solution is approximately 13.0.
Ferrous fumarate (170g, 1 mole) is slowly added to the sodium hydroxide solution at room temperature with gentle stirring.
Maltol (408g, 3.23 moles) is added to a separate volume of sodium hydroxide (40g, 1 mole) dissolved in water (100 ml) and dissolved. The pH of the solution is approximately 11.
The ferrous fumarate solution is slowly added to the maltol solution with gentle stirring. A deep red precipitate forms; the supernatant is a deep red colour.
The solution is slowly evaporated to dryness at 60 to 80° C until the material is suitable for powdering. The material is powdered and the powder is then dried to a constant weight. The yield of the final product is 615g.
The final product comprises ferric trimaltol and sodium fumarate.
EXAMPLE 3
Synthesis of ferric trimaltol using sodium carbonate to vary pH
Sodium carbonate (2.5g) is dissolved in 10ml of distilled water at room temperature. The pH of the solution is 11.6. Maltol (9.6g – three molar equivalents of sodium carbonate) is added to the sodium carbonate solution to give a cream coloured solution having a pH of 10.0.
A stoichiometric amount of ferric citrate (5g, allowing for a small excess of maltol) in an aqueous solution of sodium hydroxide (lg in 5ml of distilled water) is added slowly to the solution of maltol. The pH of the combined solutions is about 9. A red precipitate appears which is separated by decantation and dried at 80°C in an oven.
The red precipitate is ferric trimaltol, as confirmed by UV-Vis spectrometry.
EXAMPLE 4
Synthesis of ferric trimaltol using ferrous gluconate
Potassium hydroxide (5.5g) is dissolved in 50ml of distilled water at room temperature. To 25ml of this solution, maltol (16.5g, 0.13 moles) is added and gently heated to form a clear solution. To the other 25ml aliquot of the potassium hydroxide solution ferrous gluconate (22.5g) is added. This is gently heated to form a dark green saturated solution. The ferrous gluconate solution is added to the maltol solution and immediately a colour change to dark brown is noted.
On cooling, a deep brown precipitate forms (which is ferric trimaltol). The supernatant is a deep brown solution containing ferric trimaltol and potassium gluconate. The precipitate and the supernatant are dried separately at 80°C in an oven. The ferric trimaltol is a deep red brown powder with a characteristic caramel odour and UV-vis spectrum in aqueous solution.
EXAMPLE 5
Synthesis of ferric trimaltol using solid ferrous gluconate
Example 4 was repeated with the modification that the maltol is added to all of the 50 ml solution of potassium hydroxide and then solid ferrous gluconate is added directly to the maltol solution. This method gives similar end products to Example 4.
EXAMPLE 6
Synthesis of ferric trimaltol using sodium ferrous citrate
A 20% solution w/v of sodium ferrous citrate in distilled water is prepared from 7.5g of sodium ferrous citrate in 37.5ml of water. The solution of sodium ferrous citrate is dark green with an iron content of about 20%. A solution of maltol (containing 10g/50ml) in 20% sodium hydroxide is added to the solution of sodium ferrous citrate. A characteristic deep red/brown iron complex of ferric trimaltol is formed.
EXAMPLE 7
Synthesis of ferric trimaltol using solid sodium ferrous citrate
Example 6 was repeated using the same amounts and concentrations of components but the method is varied in that solid sodium ferrous citrate (7.5g) is added directly to the maltol solution (containing lOg of maltol in 50ml). Ferric trimaltol is formed using this alternative method.
EXAMPLE 8
Synthesis of ferric trimaltol using sodium ferric citrate
A 20% solution w/v of sodium ferric citrate in distilled water is prepared from 7.5g of sodium ferric citrate in 37.5ml of water. The solution of sodium ferric citrate is dark brown with an iron content of about 20%.
A solution of maltol (containing 10g/50ml) in 20% sodium hydroxide is added to the solution of sodium ferric citrate. A characteristic deep red/brown iron complex of ferric trimaltol is formed. EXAMPLE 9
Example 8 was repeated using the same amounts and concentrations of components but the method is varied in that solid sodium ferric citrate (7.5g) is added directly to the maltol solution (containing lOg of maltol in 50ml). Ferric trimaltol is formed using this alternative method.
If any of Examples 3 to 9 are repeated using maltol in a neutral or acidic aqueous medium, such as for example in buffered citric acid, brown/black impurities appear and insoluble fractions are formed (probably of ferric hydroxide) and the UN-vis spectra of the solutions are not correct. In particular, there is a peak shift towards 510nm indicating the formation of mono or dimaltol complexes or compounds.
PATENT
WO 2017167970
POLYMORPH
GB 2531742
PATENT
WO 2016066555
https://patents.google.com/patent/WO2016066555A1/en
An adequate supply of iron to the body is an essential requirement for tissue growth and the maintenance of good health in both man and animals. Moreover, in certain pathological conditions where there is an insidious blood loss, or where there is a mal-distribution of iron in the body, there may be a state of low iron stores in the body leading to an iron deficiency and a concomitant chronic anaemia. This is seen in inflammatory diseases of the gastrointestinal tract, such as gastric and peptic ulcers, reflux oesophagitis, ulcerative colitis and Crohn’s disease.
Anaemia can also follow operations that result in serious blood loss and can be associated with gastrointestinal infections, such as those caused by Helicobacter pylori.
Ferric maltol comprises a complex of one ferric iron and three maltol anions and has the following molecular formula: (C6H503)3Fe. Maltol is also known as 3-hydroxy-2-methyl-4- pyrone.
Polymorphic forms occur where the same composition of matter crystallises in a different lattice arrangement, resulting in different thermodynamic properties and stabilities specific to the particular polymorphic form. WO 03/097627 A1 discloses a method of forming iron hydroxypyrone compounds.
EP 0 159 917 A3 describes a pharmaceutical composition containing a hydroxypyrone-iron complex. WO 2012/101442 A1 discloses a method of forming iron hydroxypyrone compounds.
Schlindwein et al (Dalton Transactions, 2006, Vol. 10, pages 1313-1321) describes lipophilic 3-hydroxy-4-pyridinonate iron(lll) complexes. Ferric maltol has been known for about 100 years but no polymorphs have been identified or studied prior to this invention.
We have now found that it is possible to produce different polymorphs of ferric maltol, which crystalline forms may be referred to herein as the “compounds of the invention”. One polymorph form can be preferable in some circumstances when certain aspects, such as ease of preparation and stability, such as thermodynamic stability are required. In other situations, a different polymorph may be preferred for greater solubility and/or superior pharmacokinetics. The polymorphs of the invention can provide advantages in terms of improved or better bioavailability or improved or better stability or solubility.
The term “ferric maltol” as used herein refers to both ferric trimaltol and the designation INN ferric maltol. In one aspect of the invention there is provided a Form I polymorph of ferric maltol characterized by a powder X-ray diffraction pattern comprising characteristic crystalline peaks expressed in degrees 2-theta at each of 15.6 and 22.5 ± 0.25 or 0.2 degrees, optionally wherein the Form I polymorph comprises greater than about 92 wt.% ferric maltol based on the weight of the polymorph, such as greater than about 95 wt.%, preferably greater than about 96 wt.%, or about 98 wt.%, or about 99 wt.% such as about 99.8 wt.%.
In a further aspect of the invention there is provided a Form II polymorph of ferric maltol characterized by a powder X-ray diffraction pattern comprising a peak expressed in degrees 2-theta at 8.3 ± 0.25 degrees.
In a yet further aspect of the invention there is provided a Form III polymorph of ferric maltol characterized by a powder X-ray diffraction pattern comprising a peak expressed in degrees 2-theta at 7.4 ± 0.25 degrees. In a still further aspect of the invention there is provided a Form IV polymorph of ferric maltol characterized by a powder X-ray diffraction pattern comprising peaks expressed in degrees 2-theta at 9.5 and 14.5 ± 0.2 degrees.
The measurements of degrees 2-theta generally refer to measurements at ambient temperature, such as from about 5 to about 40°C, preferably about 10 to about 30°C. The relative intensities of the peaks can vary, depending on the sample preparation technique, the sample mounting procedure, the particular instrument employed, and the morphology of the sample. Moreover, instrument variation and other factors can affect the 2-theta values. Therefore, XRPD peak assignments for the polymorphs of the invention, as defined herein in any embodiment, can vary by, for example, ± 0.2, such as ±0.1 or ±0.05. The term “about” in relation to XRPD peak values may include for example, ±0.25 or ± 0.2, such as ±0.1 or ±0.05. These ranges may apply to any of the peak values in degrees referred to herein.
In another embodiment of the invention, there is provided a process for the preparation of a ferric maltol polymorph, such as Form I or Form II polymorph, which comprises combining ferric citrate with maltol anions to form a mixture comprising ferric maltol and wherein the process comprises the use of a ferric maltol seed crystal. The seed crystal may comprise a Form I and/or Form II polymorph as described herein and these polymorphs may be prepared using the methods described herein.
In another aspect of the invention, there is provided a process for the preparation of Form I polymorph, which comprises combining ferric citrate with maltol anions to form a mixture comprising ferric maltol polymorph Form I wherein the process comprises the use of a ferric maltol seed crystal comprising Form I and/or Form II polymorph and preferably wherein the polymorph formed is washed (typically with water) prior to drying.
In a further aspect of the invention, there is provided a process for the preparation of Form II polymorph, which comprises combining ferric citrate with maltol anions in solution to form a mixture comprising ferric maltol polymorph Form II, wherein the process preferably comprises the use of a ferric maltol seed crystal comprising Form I and/or Form II polymorph and preferably wherein the polymorph formed is washed (typically with water) prior to drying.
The invention also provides a pharmaceutical composition comprising a polymorph according to the invention, or mixtures thereof, and a pharmaceutically acceptable adjuvant, diluent or carrier. In addition, the invention provides a composition comprising Form I and Form II polymorphs as defined herein.
In an aspect of the invention, the polymorph of the invention is for use in the prevention or treatment of iron deficiency with or without anaemia in a subject. The anaemia is preferably iron deficiency anaemia.
In a further aspect of the invention there is provided the use of a polymorph of the invention for the manufacture of a medicament for the prevention or treatment of iron deficiency with or without anaemia in a subject. The anaemia is preferably iron deficiency anaemia.
The invention further provides a method for the prevention or treatment of iron deficiency with or without anaemia which method comprises the administration of a polymorph according to the invention to a subject in need of such treatment. The anaemia is preferably iron deficiency anaemia.
Preferably the polymorphs of the invention are obtained in forms that are greater than about 90%, such as greater than about 95%, crystalline (e.g. greater than about 98% crystalline and, particularly, 100%, or nearly 100%, crystalline). By “substantially crystalline” we include greater than about 60%, preferably greater than about 75%, and more preferably greater than about 80% (such as about 90%) crystalline. The degree (%) of crystallinity may be determined by the skilled person using X-ray powder diffraction (XRPD). Other techniques, such as solid state NMR, FT-IR, Raman spectroscopy, differential scanning calorimetry (DSC) microcalorimetry and calculations of the true density may also be used.
The polymorphs of the invention may be characterised by an X-ray powder diffraction pattern comprising the following characteristic crystalline peaks with approximate 2-Theta values (in degrees) as well as an indication of the relative intensity of those peaks in brackets, where a percentage relative intensity of approximately 25- 00% is termed “vs” (very strong), approximately 10-25% is termed “s” (strong), approximately 3-10% is termed “m” (medium) and approximately 1-3% is termed “w” (weak).
Form I: The Form I polymorph preferably comprises characteristic crystalline peaks with 2-Theta values (in degrees) of around (i.e. at about or at approximately) 15.6 and 22.5 ± 0.25, or 0.2 degrees. The diffraction pattern typically does not comprise peaks at one or more, or all, or each of, about 6.9, 7.4, 8.3, 9.3, 10.5, or about 11.8 degrees, such as 8.3 or 11.8 ± 0.25, or ± 0.2, or ±0.1 such as about ±0.05 degrees.
Form II:
The form II polymorph preferably comprises a characteristic crystalline peak with 2-Theta value (in degrees) of around (i.e. at about or at approximately) 8.3 ± 0.25, or ± 0.2, or +0.1 such as about ±0.05 degrees. The diffraction pattern typically does not comprise peaks at one or more, or all, or each of, about 6.9, 7.4, 9.3, 9.5, 10.5, 11.4 or about 13.7 degrees, such as 11.4 or 13.8 ±0.25, or ±0.2, or ±0.1 such as about ±0.05 degrees.
The Form III polymorph preferably comprises a characteristic crystalline peak with 2-Theta value (in degrees) of around (i.e. at about or at approximately) 7.4 ±0.3, ±0.25, or 0.2, or ±0.1 such as about ±0.05 degrees. The diffraction pattern typically does not comprise peaks at one or more, or two or more, or three or more or each of, about 6.9, 8.3, 9.5, 11.3, 12.0, 12.5, 12.9, 14.5, or about 15.8 degrees, such as 6.9, 9.5, 11.3 ±0.25, or ±0.2, or ±0.1 such as about ±0.05 degrees.
The form IV polymorph preferably comprises a characteristic crystalline peaks with 2-Theta values (in degrees) of around (i.e. at about or at approximately) 9.5 and 14.5 +0.2, or ±0.1 such as about ±0.05 degrees. The diffraction pattern typically does not comprise peaks at one or more, or two or more, or three or more or each of, about 6.9, 8.3, 10.5, 11.7, 12.0, 12.2, 12.5, 13.0, 13.4, and about 15.8 degrees, such as 6.9, 8.3, 11.7 ±0.25, or ±0.2, or ±0.1 such as about ±0.05 degrees.
Example 1 : Form I 9.04 kg ferric citrate was combined with 29 litres of purified water. Separately, 12.2 kg of maltol was combined with 15.2 litres of sodium hydroxide solution (20 % w/w). The ferric citrate and sodium hydroxide were charged into a vessel with the addition of 4 litres of water and then stirred at 20 to 25°C. A seed was then added. The seed was 65g of ferric maltol polymorph in 12 litres of water. The seed crystal was prepared by the same process as described in Example 1 but without the use of a seed crystal. The seed was added to the vessel to aid a consistent crystallisation/precipitation. The mixture was held in the vessel, as a suspension, to allow crystal growth and then filtered and washed three times, each time with 13 litres of water. The resulting solid was dried at less than 80°C and produced 13.25 kg of dried ferric maltol.
The ferric maltol in Example 1 was produced on a scale of 12 to 15 kg in different batches. The analysis of the ferric maltol produced showed the % w/w of iron present was about 12.8 to 13.0 and the % w/w of maltol present was about 87.6 to 89.3.
Patent
//////////////Ferric Maltol, マルトール第二鉄 , Feraccru, FDA 2019
CC1=C(C(=O)C=CO1)[O-].CC1=C(C(=O)C=CO1)[O-].CC1=C(C(=O)C=CO1)[O-].[Fe+3]