














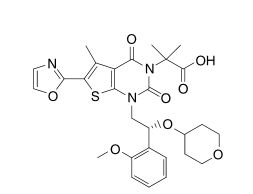
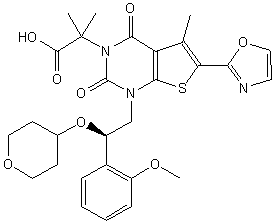
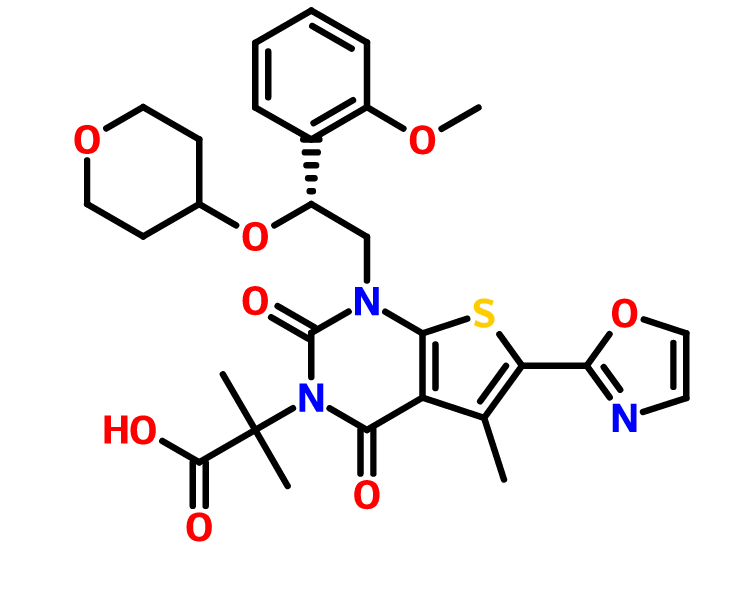
CAS: 1434635-54-7, UNII: XE10NJQ95M
PHASE 2, Non-alcoholic steatohepatitis, GILEAD
| Molecular Formula: | C28H31N3O8S |
|---|---|
| Molecular Weight: | 569.62604 g/mol |

| Company | Nimbus Therapeutics LLC |
| Description | Small molecule allosteric inhibitor of acetyl-coenzyme A carboxylase alpha (ACACA; ACC1) and acetyl-coenzyme A carboxylase beta (ACACB; ACC2) |
| Molecular Target | Acetyl-Coenzyme A carboxylase alpha (ACACA) (ACC1) ; Acetyl-Coenzyme A carboxylase beta (ACACB) (ACC2) |
| Mechanism of Action | Acetyl-coenzyme A carboxylase alpha (ACACA) (ACC1) inhibitor; Acetyl-coenzyme A carboxylase beta (ACACB) (ACC2) inhibitor |
| Therapeutic Modality | Small molecule |
1,4-Dihydro-1-((2R)-2-(2-methoxyphenyl)-2-((tetrahydro-2H-pyran-4-yl)oxy)ethyl)-alpha,alpha,5-trimethyl-6-(2-oxazolyl)-2,4-dioxothieno(2,3-d)pyrimidine-3(2H)-acetic acid
In April 2016, Gilead Sciences and Nimbus Therapeutics, LLC announced that the companies have signed a definitive agreement under which Gilead will acquire Nimbus Apollo, Inc., a wholly-owned subsidiary of Nimbus Therapeutics, and its Acetyl-CoA Carboxylase (ACC) inhibitor program. Nimbus Therapeutics will receive an upfront payment of $400 million, with the potential to receive an additional $800 million in development-related milestones over time.
The Nimbus Apollo program includes the lead candidate NDI-010976, an ACC inhibitor, and other preclinical ACC inhibitors for the treatment of non-alcoholic steatohepatitis (NASH), and for the potential treatment of hepatocellular carcinoma (HCC) and other diseases.
In May 2016, Nimbus Therapeutics announced the recent closing of Gileads acquisition of Nimbus Apollo. The acquisitions completion triggered a $400 million upfront payment to Nimbus from Gilead.
In January 2016, fast track designation was assigned in the U.S. for this indication. In May 2016, Gilead Sciences acquired Nimbus Apollo from Nimbus Therapeutics, including its acetyl-CoA carboxylase (ACC) inhibitor program.
Gilead Sciences following the acquisition of Nimbus Apollo , is developing firsocostat , the lead from a program of acetyl-CoA carboxylase (ACC)-targeting compounds, for treating fatty liver disease including non-alcoholic steatohepatitis.
Acetyl CoA carboxylase 1/2 allosteric inhibitors – Nimbus
Therapeutics
The Liver Meeting 2015 – American Association for the Study of Liver Diseases (AASLD) – 2015 Annual Meeting, San Francisco, CA, USA
Nimbus compounds targeting liver disease in rat models
Data were presented by Geraldine Harriman, from Nimbus Therapeutics, from rat models using acetyl-CoA carboxylase (ACC) inhibitors NDI-010976 (ND-630) and N-654, which improved metabolic syndrome endpoints, decreased liver steatosis, decreased expression of inflammatory markers and improved fibrosis. The hepatotropic ACC inhibitor NDI-010976 had IC50 values of 2 and 7 nM for ACC1 and 2, respectively, EC50 values in HepG2 serum free and 10% serum of 9 and 66 nM, respectively, and 2-fold C2C12 fatty acid oxidation (FAOxn) stimulation at 200 nM. Rat FASyn (synthase), malonyl-CoA (liver) and malonyl-COA (muscle) respective ED50 values were 0.14 mg/kg po, 0.8 and 3 mg/kg. The rat respiratory quotient (RQ) MED was 3 mg/kg po. ADME data showed low multispecies intrinsic clearance (human, mouse, rat, dog, monkey). NDI-010976 was eliminated predominantly as the parent drug. Additionally, P450 inhibition was > 50 microM. In liver and muscle, NDI-010976 modulated key metabolic parameters including a dose-dependent reduction in the formation of the enzymatic product of acetyl coA carboxyloase malonyl coA; the ED50 value was lower in muscle. The drug also decreased FASyn dose dependently and increased fatty acid oxidation in the liver (EC50 = 0.14 mg/kg). In 28-day HS DIO rats, NDI-010976 favorably modulated key plasma and liver lipids, including decreasing liver free fatty acid, plasma triglycerides and plasma cholesterol; this effect was also seen in 37-day ZDF rats

http://www.google.com/patents/WO2013071169A1?cl=en
Example 76: Synthesis of 2-[l-[2-(2-methoxyphenyl)-2-(oxan-4-yloxy)ethyl]-5- methyl-6-(l,3-oxazol-2-yl)-2,4-dioxo-lH,2H,3H,4H-thieno[2,3-d]pyrimidin-3-yl]-2- methylpropanoic acid (1-181).

Synthesis of compound 76.1. Into a 250-mL 3 -necked round-bottom flask, purged and maintained with an inert atmosphere of nitrogen, was placed oxan-4-ol (86 g, 842.05 mmol, 2.01 equiv) and FeCl3 (10 g). This was followed by the addition of 57.2 (63 g, 419.51 mmol, 1.00 equiv) dropwise with stirring at 0 °C. The resulting solution was stirred for 3 h at room temperature. The resulting solution was diluted with 500 mL of H20. The resulting solution was extracted with 3×1000 mL of ethyl acetate and the organic layers combined. The resulting solution was extracted with 3×300 mL of sodium chloride (sat.) and the organic layers combined and dried over anhydrous sodium sulfate. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1 : 10). This resulted in 22 g (21%) of 76.1 as a white solid.
Synthesis of compound 76.2. The enantiomers of 76.1 (22g) were resolved by chiral preparative HPLC under the following conditions (Gilson Gx 281): Column: Venusil Chiral OD-
H, 21.1 *25 cm, 5 μιη; mobile phase: hexanes (0.2% TEA) and ethanol (0.2% TEA) (hold at 10% ethanol (0.2%TEA) for 13 min); detector: UV 220/254 nm. 11.4 g (52%) of 76.2 were obtained as a white solid.
Synthesis of compound 76.3. Into a 500-mL 3-necked round-bottom flask, purged and maintained with an inert atmosphere of nitrogen, was placed 70.1 (12 g, 20.49 mmol, 1.00 equiv), tetrahydrofuran (200 mL), 76.2 (6.2 g, 24.57 mmol, 1.20 equiv) and DIAD (6.5 g, 32.18 mmol, 1.57 equiv). This was followed by the addition of a solution of triphenylphosphane (8.4 g, 32.03 mmol, 1.56 equiv) in tetrahydrofuran (100 mL) dropwise with stirring at 0 °C in 60 min. The resulting solution was stirred overnight at room temperature. The resulting mixture was concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1 :5). This resulted in 17 g (crude) of 76.3 as a white solid.
Synthesis of compound 76.4. Into a 500-mL 3-necked round-bottom flask, purged and maintained with an inert atmosphere of nitrogen, was placed 76.3 (17 g, crude), toluene (300 mL), Pd(PPh3)4 (1.7 g, 1.47 mmol, 0.07 equiv) and 2-(tributylstannyl)-l,3-oxazole (8.6 g, 24.02 mmol, 1.16 equiv). The resulting solution was stirred overnight at 110 °C. The resulting mixture was concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1 : 10). Purification afforded 6 g of 76.4 as a white solid.
Synthesis of compound 1-181. Into a 250-mL 3-necked round-bottom flask, was placed 76.4 (6 g, 7.43 mmol, 1.00 equiv), tetrahydrofuran (100 mL), TBAF (2.3 g, 8.80 mmol,
I .18 equiv). The resulting solution was stirred for 1 h at room temperature. The resulting mixture was concentrated under vacuum. The residue was applied onto a silica gel column with dichloromethane/methanol (50: 1). This resulted in 3.4 g (80%) of Compound 1-181 as a white solid.
Purification: MS (ES): m/z 570 (M+H)+, 592 (M+Na)+.
1H NMR (300 MHz, DMSO- d6): δ 1.22-1.36 (m, 2H), 1.62 (m, 8H), 2.75 (s, 3H), 3.20-3.39 (m, 3H), 3.48-3.58 (m, 2H), 3.80 (s, 3H), 3.85-4.20 (m, 2H), 5.30 (m, 1H), 7.03 (m, 2H), 7.33-7.50 (m, 3H), 8.2 (s, 1H).
Conference: 66th Annual Meeting of the American Association for the Study of Liver Diseases Conference Start Date: 13-Nov-2015
…candidates for minimizing IR injury in liver transplantation.Nimbus compounds targeting liver disease in rat modelsData were presented by Geraldine Harriman, from Nimbus Therapeutics, from rat models using acetyl-CoA carboxylase (ACC) inhibitors NDI-010976 (ND–630) and N-654, which improved metabolic syndrome endpoints, decreased liver steatosis, decreased expression of inflammatory markers and improved fibrosis. The hepatotropic ACC inhibitor NDI-010976 had IC50 values of 2 and 7 nM for ACC1 and 2, respectively…
REFERENCES
The Liver Meeting 2015 – American Association for the Study of Liver Diseases (AASLD) – 2015 Annual Meeting San Francisco, CA, USA ,
| WO-2014182943 |
| WO-2014182945 |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015203510 | 2015-07-23 | ACC INHIBITORS AND USES THEREOF |
| US2013123231 | 2013-05-16 | ACC INHIBITORS AND USES THEREOF |
WO2017151816 ,
CN 107629069
CN 107629069
CN 107151251
WO 2013071169
WO 2016112305
PATENT
WO-2018161022
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018161022&tab=PCTDESCRIPTION&maxRec=1000
Solid forms, including a salts (such as choline, diethylamine, NN-dibenzylethylenediamine, ethanolamine) or co-crystal, of firsocostat and compositions comprising them are claimed, which exhibits Acetyl-CoA carboxylase inhibitory activity and useful for treating ACC mediated diseases such as metabolic disorders, neurological disorders, and infectious diseases. Also claimed are process for preparing firsocostat and intermediates useful for preparing them are claimed.
The present disclosure provides forms of Compound I or a compound of formula (I) having the formula:
Compound I may be referred to by formula (I):
(I)
or its chemical name of (R)-2-(l-(2-(2-methoxyphenyl)-2-((tetrahydro-2H-pyran-4-yl)oxy)ethyl)-5-methyl-6-(oxazol-2-yl)-2,4-dioxo-l,2-dihydrothieno[2,3-d]pyrimidin-3(4H)-yl)-2-methylpropanoic acid. U.S. Patent No. 8,969,557 discloses that Compound I exhibits ACC inhibitory activity. In the present disclosure, compounds may be presented in the form of chemical structures or names.
Scheme 1 represents an exemplary synthesis of a compound of formula (F) and may be carried out according to the embodiments described herein.
Scheme 1
(E) (F)
Scheme 2
(E-1 ) (I)
Scheme 3
Step (g)
Scheme 4
scheme 5
Example 1 : Synthesis of Compound B-2
B-2
[0401] Compound A-2 was combined with Compound G-1 (about 1 equivalents (“equiv”)) with K2CO3 (about 2.3 equiv) in dimethylacetamide. The mixture was stirred at room temperature. The resulting mixture was then diluted with ethyl acetate and washed with water and brine. The organic layer was separated and concentrated to dryness, and the resulting product was purified by column chromatography (eluent: 0 to about 28% ethyl acetate:
heptanes). The resulting product was Compound B-2. ¾ NMR (300 MHz, CDCh): δ 7.92 (d, J
= 8.4 Hz, 1H), 7.57 (m, 1H), 7.06 (m, 2H), 5.20 (s, 2H), 4.00 (s, 3H), 2.42 (s, 3H), 1.77 (s, 6H), 1.44 (s, 9H).
Example 2: Synthesis of a compound of formula (C)
(B) (C)
[0402] Compound of formula (B) or Compound B (which may be prepared as described in Example 1) and a (S,S)-Ruthenium catalyst, such as a Ruthenium catalyst as described herein, or a suitable antipode of the Ruthenium catalyst, are combined in the presence of potassium tert-butoxide (“KO^-Bu”) and isopropanol and refluxed to yield a compound of formula (C) or Compound C. Compound C is isolated and purified by methods described herein.
Example 3: Synthesis of Compound D-1
C-1 D-1
[0403] To Compound C-1 in dichloromethane is added 4-bromotetrahydro-2H-pyran. Upon addition of an organic base, the reaction mixture is stirred ovemight to yield a compound of formula D-1 or Compound D-1. Compound D-1 is isolated and purified by the methods described herein.
Example 4: S
D-1 E-2
[0404] Oxazole in THF is cooled to between about -80 °C and about -60 °C. Then, ft-butyllithium in hexanes is added while maintaining the temperature of the reaction below about -60 °C. The mixture is stirred at this temperature for 90 minutes. Zinc (II) chloride is added, maintaining the temperature of the mixture below about -60 °C, and the mixture is stirred at that temperature for about one hour before warming to about 10-20 °C. Compound D-1 is added to the reactor followed by tetrakis(triphenylphosphine)palladium(0) (“Pd(PPh3)4”), and the temperature is adjusted to between about 55-65 °C. The mixture is stirred at that temperature for about 12 hours to yield Compound E-2. Compound E-2 is isolated and purified by the methods described herein.
Example 5: Synthesis of Compound I
[0405] A sulfuric acid solution was prepared by addition of concentrated sulfuric acid (47 g,
4.7 w/w Compound E-2) to water (12 g, 1.2 v/w Compound E-2) followed by a water (15 g, 1.5 v/w Compound E-2) rinse forward. 2-Propanol (37 g, 4.7 v/w Compound E-2) was slowly charged to a reactor containing sulfuric acid solution at about 9 °C while maintaining the reaction contents at no more than about 40 °C, and the solution was cooled to about 5 °C .
Compound E-2 (10 g, 1.0 equiv) was charged to the solution, followed by a 2-propanol rinse forward (2 g, 0.25 v/w E-2). The contents were cooled to about 7 °C and stirred for a minimum of about 21 hours. The contents were slowly added into water, and the slurry was agitated for about 30 minutes. The slurry was filtered, and the filter cake was washed and dried under vacuum for about 4 hours. The crude wet cake was charged back to the reactor, followed by additions of ethyl acetate (40 g, 4.4 v/w Compound E-2) and water (100 g, 10 v/w Compound E-2). The slurry was adjusted to pH at about 8-9 with an about 20 wt% sodium hydroxide solution at about 22 °C, and then agitated for about 30 minutes at about 22 °C. The solution was allowed to settle. The top organic layer was collected and the bottom aqueous layer was washed with ethyl acetate (40 g, 4.4 v/w Compound E-2) at about 22 °C for about 30 minutes. The solution was allowed to settle, and the top organic layer was removed. 2-Methyltetrahydrofuran (86 g, 10 v/w Compound E-2) was then added, was adjusted to pH at about 4-5 with an about 4 N HCl solution at about 22 °C. The solution was agitated for about 30 minutes at about 22 °C and then allowed to settle. The bottom aqueous layer was extracted with 2-methyltetrahydrofuran (52 g, 6 v/w Compound E-2) at about 22 °C for about 30 minutes. After the solution was allowed to settle, the bottom aqueous layer was removed. The organic layers were combined and distilled under vacuum (jacket at about < 45 °C) to about 4V pot volume. Ethanol (55.4 g, 7 v/w
Compound E-2) was added and the reaction as distilled (repeated twice). Ethanol was again added (23.7 g,3 v/w Compound E-2), followed by water (30 g, 3 v/w Compound E-2). The reaction was heated to about 75 °C and then cooled over about 4 hours to about 50 °C, then to about 0 °C over about 5 hours. The reaction was then aged and filtered, and the solid was washed with a precooled mixture of ethanol (9.5 g, 1.2 v/w Compound E-2) and water (6 g, 0.6 v/w Compound E-2). The resulting product was washed to afford Compound of formula (I). ¾ NMR (400 MHz, CDCh): δ 7.70 (s, 1H), 7.57 (dd, J= 1.6 Hz, J= 7.6 Hz, 1H), 7.29 (td, J= 1.6 Hz, J = 8.0 Hz, 1H), 7.23 (d, J= 0.4 Hz, 1H), 7.02 (t, J= 7.6 Hz, 1H), 6.86 (d, J= 8.4 Hz, 1H), 5.39 (dd, J= 5.6 Hz, J= 8.0 Hz, 1H), 4.17-4.14 (m, 1H), 4.04 (br, 1H), 3.86 (s, 3H), 3.78-3.67 (m, 2H), 3.46-3.40 (m, 1H), 3.37-3.32 (m, 2H), 2.85 (s, 3H), 1.87 (s, 3H), 1.83 (s, 3H), 1.75-1.72 (m, 2H), 1.59-1.51 (m, 1H), 1.48-1.39 (m, 1H).
Example 6: Synthesis of Compound J-l
Step (a): Formation of Compound P-l
[0406] 2-Methoxyphenylmagnesium bromide (1 M in THF, 1.0 equiv.) was added to a solution of diethyl oxalate (1.1 equiv.) in THF (250 mL) at about -20 °C over approximately 20 min. After aging for about 45 min at about -20 °C, the resulting slurry was quenched with saturated NH4CI (250 mL) and was diluted with water (200 mL). This mixture was extracted with EtO Ac (400 mL), and the organic phase was washed with brine (200 mL). The organic phase was concentrated and the solvent was exchanged to THF. The resulting THF solution was used in the next step as is. ¾ NMR (400 MHz, CDCh): δ 7.90 (m, 1H), 7.61 (m, 1H), 7.10 (t, J = 7.6 Hz, 1H), 7.01 (d, J= 8.4 Hz 1H), 4.41 (q, J= 7.1 Hz, 2H), 3.88 (s, 3H), 1.41 (t, J= 7.1 Hz, 3H).
Alternate Preparation Compound P-l:
[0407] Anisole (1.0 equiv.) in THF (15 mL) was cooled to about -20 °C, and 2.5 M n-BuLi/hexane (1.1 equiv.) was added. The mixture was allowed to warm to about 0 °C and aged for about 2 hours, then warmed to room temperature overnight. The solution was then added to a solution of diethyl oxalate (4.0 equiv.) in THF (10 mL) at about -20 °C. The mixture was allowed to warm to about room temperature and aged for approximately 2 hours, then cooled to about 0 °C and quenched via addition of saturated NH4CI (30 mL). This mixture was extracted with EtOAc, and the organic phase was washed with brine and dried over MgSCk
Concentration afforded Compound P-1.
Alternate Preparation Compound P-1:
[0408] 2-Bromoanisole (1.0 equiv.) in THF (63 mL) was cooled to about -65 °C and 2.5M ft-BuLi/hexanes (1.0 equiv) was added. After aging for approximately 1 h, diethyl oxalate (4.0 equiv.) was charged, and the reaction mixture was allowed to warm to about room temperature. After approximately 1 h at about room temperature, the reaction mixture was cooled to about 0 °C, quenched by addition of saturated NH4CI (50 mL), and diluted with EtOAc. The aqueous phase was separated and was extracted with EtOAc. The combined organic phases were washed with brine and dried over MgS04. Concentration under high vacuum afforded a product that was passed through a plug of silica gel to afford Compound P-1.
Step (b): Hydrolysis of Compound P-1 and salt conversion to Compound O-l:
P-1 0-1
[0409] The resulting solution of ketoester, compound P-1, in THF (about 1.0 equiv.) was cooled over an ice bath and 2N NaOH (1.36 equiv.) was added. The reaction was agitated at about 0 °C and after reaction completion, the reaction was then acidified by addition of 6N HC1 (57 mL) to about pH<l and extracted with EtOAc (500 mL). The organic phase was washed with brine (200 mL). The organic phase was concentrated and then solvent exchanged to EtOAc. The resulting solution was cooled to about 0 °C and solid KOlBu (1.0 equiv.). The slurry was agitated for approximately 4 h and the solids were filtered, rinsed with EtOAc, and dried overnight at about 60 °C under vacuum to afford Compound O-l . ¾ NMR (400 MHz, DMSO-d6): 5 7.61 (d, J= 7.6 Hz, 1H), 7.49 – 7.41 (m, 1H), 7.04 (d, J= 8.4 Hz 1H), 6.96 (t, J = 7.4 Hz, 1H), 3.73 (s, 3H).
Step (c): Reduction of Compound O-l to Compound N-1:
0-1 N-1
[0410] To triethylamine (3.6 equiv.) precooled to about 0 °C, was added formic acid (9.0 equiv.) over about 30 min while maintaining a temperature less than about 30 °C. Solid RuCl (i?,i?)-Ts-DENEB catalyst (0.07 mol%) followed by ketoacid potassium salt (1.0 equiv.) were then charged to the mixture of triethylarnine/forrnic acid. The resulting slurry was warmed to about 50 °C and was stirred under nitrogen until the reaction was complete. The reaction was cooled over an ice bath and quenched by the addition of water (76 mL) followed by 10N NaOH (128 mL) to pH>13. Water (30 mL) and iPrAc (130 mL) were added and the organic layer was separated, and the aqueous phase was extracted with iPrAc (2 χ 130 mL). The aqueous phase was cooled and was acidified with concentrated HC1. This was extracted with iPrAc several times and the combined organic extract was concentrated and solvent exchanged to toluene, filtered hot, and then cooled to about 30 °C over approximately 2 h, aged for approximately 1 h, then filtered to afford solids that were then slurry-rinsed with toluene (50 mL) at room temperature and filtered. The wet cake was dried to afford Compound N-1. ¾ NMR (400 MHz, CDCh): δ 7.44 (d, J = 7.6 Hz, 1H), 7.40 – 7.36 (m, 1H), 7.06 (t, J = 7.6 Hz 1H), 6.98 (d, J = 8.4 Hz, 1H), 5.41 (s, 1H), 3.94 (s, 3H).
Step (d): Spiroketalization to afford Compound L-1:
N-1 L-1
[0411] Compound N-1 (1.0 equiv.), tetrahydropyran-4-one (compound M, 1.1 equiv.), and MTBE (30 mL) were sequentially charged and cooled to about 0 °C. Boron trifluoride THF complex (1.4 equiv.) was added over about 10 mins. After reaction completion, the reaction was slowly quenched with a pre-mixed solution of sodium bicarbonate (3.66 g) and water (40 mL). The solution was warmed to about 20 °C and diluted with toluene (40 mL) and stirred until dissolved. Agitation was stopped and the aqueous layer removed. The organic layer was washed with water (20 mL) and removed. The organic layer was collected and reactor rinsed forward with toluene (4 mL) to yield Compound L-1. ¾ NMR (400 MHz, CDCh): δ 7.42 – 7.38 (m, 1H), 7.32 (dd, J = 7.5, 1.5 Hz, 1H), 7.03 (t, J = 7.5 Hz, 1H), 6.98 (d, J = 8.3 Hz, 1H), 5.52 (s, 1H), 3.97 – 3.79 (m, 7H), 2.18 – 1.97 (m, 4H).
Step (e): Reduction of Compound L-1 to Compound K-l :
L-1 K-1
[0412] A stock solution of spiroketal, compound L-1, in MeTHF/MTBE (1.0 equiv.) was charged to a reactor. The solution was then distilled to about 4 volumes. MeTHF (187 mL) was charged, and distilled down to about 5 volumes. The solution was cooled to about 20 °C. DCM (90 mL) was charged and the solution was cooled to about 10 °C and tert-butyl magnesium chloride (2 M in diethyl ether) (5.0 equiv.) was added over approximately 45 mins. Following addition, the contents were cooled to about 7 °C and aged overnight at about 10 °C, then to about 0 °C. A premixed solution of HC1 (45 mL) and water (126 mL) was then slowly added. The aqueous bottom layer was drained and the aqueous layer extracted with MeTHF (93 mL). The combined organic layers were washed with water (37 mL) and the remaining organic layer was distilled down to about 4 volumes. Isopropyl acetate (181 mL) was charged and the solution reduced to about 5 volumes. The reaction was cooled to about 72 °C and heptanes (58 mL) was charged and the solution was held for about 1 hour before cooling to about 0 °C over approximately 5 hours. The slurry was agitated at about 0 °C for >12 h and then filtered, rinsed with an isopropyl acetate (9 mL) and heptanes (18 mL) mixture, followed by water (54 mL). The solids were dried to yield compound K-l. ¾ NMR (400 MHz, CDCh): δ 8.49 (br. s, 1 H), 7.42 – 7.29 (m, 2H), 6.98 (t, J= 7.4 Hz, 1H), 6.92 (d, 8.3 Hz, 1H), 5.43 (s, 1H), 3.96 (dt, J = 11.5, 4.3 Hz, 1H), 3.89 (dt, J = 11.5, 4.3 Hz, 1H), 3.85 (s, 3H), 3.67 – 3.58 (m, 1H), 3.47 – 3.30 (m, 2H), 2.03 – 1.93 (m, 1H), 1.84 – 1.75 (m, 1H), 1.75 – 1.56 (m, 2H).
Step (f): Reduction of Com ound K-l to Compound J-1:
J-1
K-1
[0413] A solution of acid, compound K-l (1.0 equiv.), in THF (90 mL) was cooled to about 0 °C and NaBH4 (1.2 equiv.) was added followed by BF3 THF complex (1.5 equiv.). The solution was warmed to about 20 °C and agitated until the reaction was deemed complete. Upon completion, MeOH (24 mL) was added to the reaction mixture after adjusting the temperature to about 5 °C, and was stirred until the gas evolution ceased. EtOAc (102 mL) was charged followed by saturated NLUClaq solution (87 mL). The agitation was stopped and the aqueous layer was removed. The organic layer was distilled down to about 3 volumes under vacuum, and then heptane (46 mL) was charged. The resulting mixture was cooled to about 0 °C and agitated at this temperature for approximately 4 h before being filtered and rinsed with heptane (3 mL). The resulting solids were dried to yield compound J-1. ¾ NMR (400 MHz, CDCh): δ 7.42 (d, J = 7.2 Hz, 1H), 7.27 (m, 1H), 6.98 (m, 1H), 6.87 (d, J = 8.4 Hz, 1H), 5.06 (dd, J = 8.4, 2.8 Hz, 1H), 3.93 (m, 2H), 3.82 (s, 3H), 3.67 (m, 1H), 3.55 – 3.46 (m, 2H), 3.41 – 3.32 (m, 2H), 2.27 (d, J = 8.0 Hz, 1H), 2.01 (m, 1H), 1.80 – 1.70 (m, 1H), 1.65 (m, 2H).
Step (g): Alternate Direct Reduction of Compound L-1 to Compound J-1:
L-1 J-1
[0414] To a solution of ketal, compound L-1 (1 equiv.), in diglyme (0.7 mL) was added NaBH4 (3.6 equiv.) followed by BF3 THF complex (4.5 equiv.). Reaction mixture was agitated for about 18 hours and was quenched by dropwise addition of MeOH (1 mL) followed by saturated Ν¾(¾ solution (1 mL). EtOAc (2 mL) was added, shaken well and the aqueous layer was removed. Organic solvent was removed under reduced pressure to obtain the crude compound J-1.
Example 7: Alternate Synthesis to Compound N-1
Step (a): Addition of hydrogen cyanide to ortho-anisaldehyde, compound U-1, to form compound T-1
[0415] To an Eppendorf tube was added ort/ro-anisaldehyde, compound U-1 (1.0 equiv), followed by 0.4 M sodium acetate buffer pH 5 (0.25 mL) and fert-butyl methyl ether (0.75 mL). The mixture was shaken using a thermomixer at about 30 °C and about 1200 rpm to ensure
complete dissolution of the aldehyde. Once this was complete acetone cyanohydrin (1.15 equiv) is added to the reaction mixture followed by hydroxynitrilase enzyme (2 mg). The Eppendorf tube was shaken in a thermomixer at about 30 °C and about 1200 rpm overnight. The Eppendorf tube was then heated to about 60 °C at about 1400 rpm for about 15 mins in order to denature the enzyme before being cooled to about 30 °C. The Eppendorf tube was then centrifuged at about 13,400 rpm for about 15 mins in order to pellet the denatured enzyme from the organic layer. The organic layer was removed and concentrated to dryness to give crude compound T-l . ¾ NMR (400 MHz, CDCh): δ 7.45 – 7.39 (m, 2H), 7.04 – 6.96 (m, 2H), 5.63 (s 1H), 3.94 (s, 3H), 3.75 (br, 1H).
Step (b): Hydrolysis of c
T-1 N-1
[0416] Before starting the reaction the following stock solutions were prepared: A solution of the crude cyanohydrin (compound T-l) in DMSO (about 100 mg/mL); a solution of 50 mM potassium phosphate (pH 7) containing 2 mM dithiothreitol (DTT); and 1 mM ethylenediamine tetraacetic acid (EDTA). To an Eppendorf tube was added nitrilase enzyme (4 mg) followed by 1.1 mL of the reaction buffer solution and 0.05 mL of the solution containing the crude cyanohydrin (about 10 mg). The Eppendorf tube was shaken in a thermomixer at about 30 °C and about 1200 rpm overnight. The Eppendorf tube was then heated to about 60 °C at about 1400 rpm for about 15 mins in order to denature the enzyme before being cooled to about 30 °C once more. The Eppendorf tube was centrifuged at about 13,400 rpm for about 15 mins in order to pellet the denatured enzyme and then separate it from the supernatant. The supernatant was either sampled directly for reverse phase UPLC or extracted with DCM for normal phase HPLC. In the case of DCM extraction, after separating the layers the organic layer was concentrated to dryness before the appropriate diluent was added for normal phase HPLC. UPLC analysis showed a peak with retention time identical to a reference standard of compound N-1.
Example 8: Alternate S nthesis to Compound N-1
P-1 V-1 N-1
Step (a): Reduction of Compound P-1 to form 2 ‘-methoxy-ethyl mandelate, Compound V-1:
P-1 V-1
[0417] The following stock solutions were made prior to the start of the reaction: a solution of starting material in DMSO (about 100 mg/ mL), NADP+ or NAD+ in 0.1M phosphate buffer (as appropriate) (2 mg/mL), glucose dehydrogenase in 0.1 M phosphate buffer (4 mg/mL), and glucose in 0.1 M phosphate buffer (20 mg/mL). To an Eppendorf tube is charged the ketoreductase enzyme (2 mg) followed by 0.25 mL of buffer solution containing NAD(P)+, 0.25 mL of buffer solution containing glucose dehydrogenase (GDH) and 0.5 mL of buffer solution containing glucose. Finally, 0.05 mL of the stock solution containing the starting material, compound P-1 in DMSO is added. The Eppendorf tube was then shaken in a thermomixer at about 30 °C and about 1200 rpm overnight. The Eppendorf tube was then heated to about 60 °C at about 1400 rpm for about 15 mins in order to denature the enzymes before being cooled to about 30 °C. The Eppendorf tube was then centrifuged at about 13,400 rpm for about 15 mins in order to pellet the denatured enzyme and the supernatant removed. This was either sampled directly for reverse phase UPLC or extracted with DCM for normal phase HPLC. In the case of DCM extraction after separating the layers the organic layer was concentrated to dryness before the appropriate diluent was added for normal phase HPLC. UPLC analysis showed a peak with retention time identical to a reference standard of the product material.
Step (b) Hydrolysis of 2 ‘-methoxy-ethyl mandelate, compound V-1, to provide compound N-1:
V-1 N-1
[0418] A solution of 2′ -methoxy-ethyl mandelate (1.0 equiv.) in EtOH (30 mL) was cooled to about 0 °C and 1.25 M NaOH (30 mL) was slowly added. Upon reaction completion, the reaction was adjusted to about pH 1 with 1M HC1 (40 mL). The mixture was extracted three times with ethyl acetate (30 mL) and the combined organics were washed with a brine solution (25 mL). The combined organic layers were dried over sodium sulfate, filtered, and the solvent removed under vacuum to provide the product. NMR data reported as above.
CLIP
https://cen.acs.org/articles/94/i39/silent-liver-disease-epidemic.html

/////// ND 630, NDI 010976, ND-630, NDI-010976, NIMBUS, GILEAD, 1434635-54-7, PHASE 2
FIRSOCOSTAT, ND 630, GS-0976, NDI-010976, FAST TRACK, CS-6509
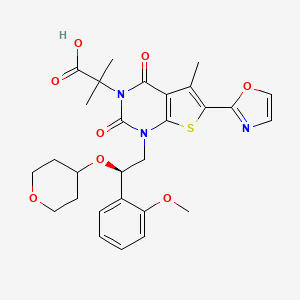
COc1ccccc1[C@H](CN2C(=O)N(C(=O)c3c(C)c(sc23)c4occn4)C(C)(C)C(=O)O)OC5CCOCC5
O=C(O)C(C)(C)N4C(=O)c1c(C)c(sc1N(C[C@H](OC2CCOCC2)c3ccccc3OC)C4=O)c5ncco5