














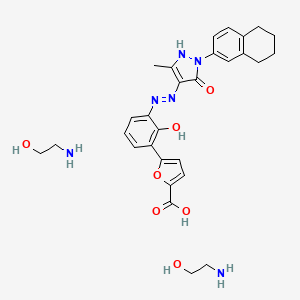
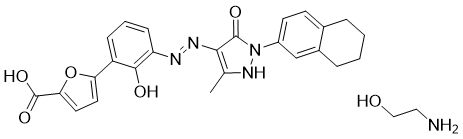
Hetrombopag Olamine, RAFUTROMBOPAG OLAMINE
- Hetrombopag diolamine
- SHR8735 olamine
- Hetrombopag ethanolamine
- SHR-8735 olamine
580.6 g/mol, C29H36N6O7, V45T2I862X
2-aminoethanol;5-[2-hydroxy-3-[[5-methyl-3-oxo-2-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazol-4-yl]diazenyl]phenyl]furan-2-carboxylic acid
CAS 1257792-42-9
1257792-41-8 (free acid) 1257792-41-8 (ethanolamine) 1257792-42-9 (olamine)
Jiangsu Hengrui Pharmaceutical, was approved in China in June 2021 for treatment of adult patients with chronic primary immune thrombocytopenia (ITP) and severe aplastic anemia who have not responded well to other treatments
Hetrombopag Olamine is the orally active ethanolamine salt of hetrombopag, a small-molecule, nonpeptide thrombopoietin receptor (TPO-R or CD110) agonist, with megakaryopoiesis-stimulating activity. Upon oral administration, hetrombopag targets, binds to and stimulates the transmembrane domain of the platelet TPO-R, a member of the hematopoietin receptor superfamily. Activation of TPO-R leads to the proliferation and differentiation of cells in the megakaryocytic lineage and an increase in platelet production. This may prevent or treat chemotherapy-induced thrombocytopenia.
- OriginatorJiangsu Hengrui Medicine Co.
- DeveloperAtridia; Jiangsu Hengrui Medicine Co.
- ClassAntianaemics; Antihaemorrhagics; Aza compounds; Carboxylic acids; Furans; Pyrazolones; Small molecules; Tetrahydronaphthalenes
- Mechanism of ActionThrombopoietin receptor agonists
- Orphan Drug StatusYes – Thrombocytopenia
- MarketedAplastic anaemia; Idiopathic thrombocytopenic purpura
- Phase IIIThrombocytopenia
- No development reportedUnspecified
- 07 Dec 2024Efficacy and adverse events data from a phase-III trial in Aplastic anaemia presented at the 66th American Society of Hematology Annual Meeting and Exposition (ASH-Hem-2024)
- 31 Jul 2024Phase-III clinical trials in Thrombocytopenia in China (PO) (NCT06507436)
- 25 Jul 2024Jiangsu Hengrui Medicine plans a phase III trial in Thrombocytopenia (PO) in July 2024 (NCT06507436)
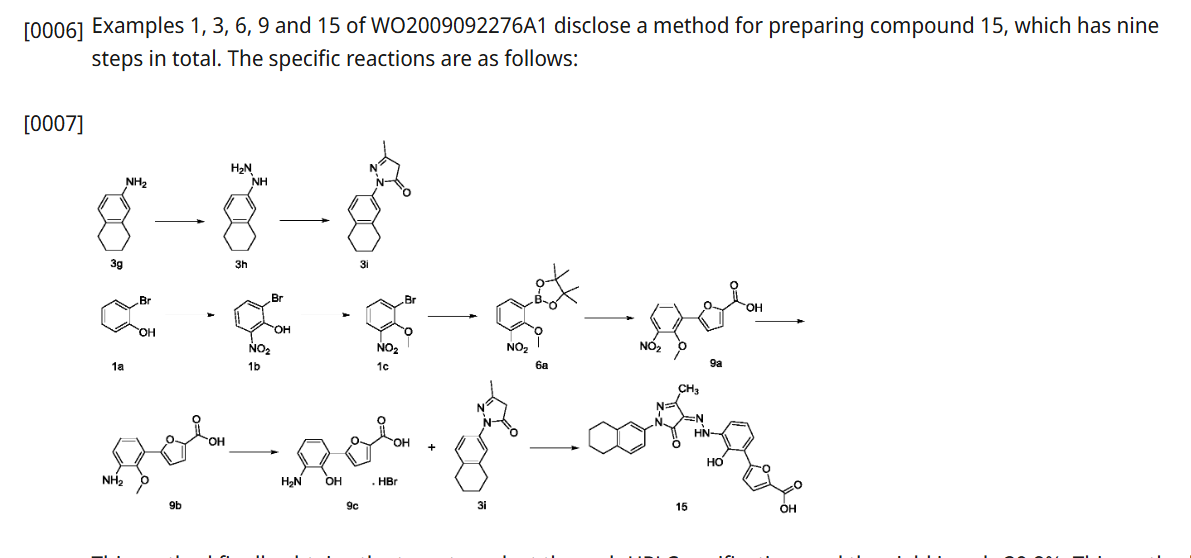
SYN
CN 113929668
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN349207982&_cid=P21-MDCUSL-44897-1
| Example 1. Synthesis of 5-(2-carbonyl-2,3-dihydrobenzoxazol-7-yl)furan-2-carboxylic acid |
| |
| Add purified water to the batching barrel, add 4.0kg of compound a under stirring, then add 10L of hydrochloric acid, stir, pump the material into a 50L reactor, add 10L of purified water to the batching barrel and pump it into the reactor. Turn on stirring, start cooling, the temperature drops to -5~2°C, start adding sodium nitrite aqueous solution (6.4L purified water, 1840g sodium nitrite), keep the temperature in the reactor no higher than 5°C during the process; after adding, continue stirring for 10~20min; add 800g of urea, continue stirring for 10~20min, the obtained diazonium salt solution is ready for use, and the temperature in the whole process is kept no higher than 5°C. |
| 44kg of acetone was pumped into a 200L reactor, and 15.0kg of compound b and 463.5g of copper chloride dihydrate were added in sequence under stirring. The temperature was raised to 30-35°C, and the obtained diazonium salt solution was added. The temperature was maintained at 30-40°C during the period. After the addition was completed, the temperature was maintained at 30-40°C and the reaction was continued with stirring for 1-1.5h. 120.0L of purified water was added, the temperature was raised to 40-45°C, and stirring was continued for a period of time. Filter, wash the filter cake with purified water until the filtrate is neutral, filter again, and collect the filter cake. 80L of purified water was added to the reactor, stirring was started, and the filter cake was added. Sodium hydroxide aqueous solution was added to the reactor to adjust the pH, the pH value was maintained at 8-10 for a period of time, and the filtrate was pumped into the reactor, and the filter was pressed into the material barrel through the filter press. Then 10L of purified water was pumped into the reactor and filtered into the material barrel. The material in the material barrel was pumped into the reactor, and then ethyl acetate was pumped in, stirred, and allowed to stand for 30-40 minutes. The aqueous phase was separated and collected, and the aqueous phase was pumped into the reactor, and the pH was adjusted to 3-4 with hydrochloric acid solution, and the filter cake was washed with purified water until the filtrate was neutral, and then the filter cake was collected. The filter cake was dried to obtain compound c. The yield of this step was 3.59 kg, and the yield was 55%. |
| Example 2: Synthesis of 5-(3-amino-2-hydroxyphenyl)furan-2-carboxylic acid |
| |
| Purified water was pumped into the 50L reactor, stirring was started, 3.53kg of sodium hydroxide was added, and compound c obtained in the previous step was added. Under nitrogen protection, the reaction mixture was heated to reflux in the reactor for reaction. After the reaction, the reaction solution was cooled, the temperature was lowered to 0-10°C, and hydrochloric acid solution was added to adjust the pH value to 5-6. The filter cake was filtered, and the filtrate was washed with purified water until neutral, and then filtered again to collect the filter cake. The filter cake was dried to obtain compound d. The yield in this step was 2.78kg, with a yield of 90%. |
| Example 3. Synthesis of (Z)-5-(2-hydroxy-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazol-4(5H)-ylidene)hydrazino)phenyl)furan-2-carboxylic acid |
| |
| Purified water was added to the batching barrel, and compound d was added in sequence under stirring, and then 6.3L hydrochloric acid was added, and the materials were pumped into a 200L reactor. Purified water was added to the batching barrel again, and then pumped into the reactor. Stirring was started, and the temperature was lowered to -5 to 2°C. Sodium nitrite aqueous solution (sodium nitrite to compound d molar ratio is 1:1) was added, and the internal temperature was kept at no more than 5°C during the process. After the addition was completed, stirring was continued; urea was added, and stirring was continued to obtain a diazonium salt solution for use, and the internal temperature was kept at no more than 5°C during the whole process. |
| Add 36L purified water and 4000g sodium hydroxide to the batching barrel, stir to dissolve, and set aside. Take 26kg of the above sodium hydroxide aqueous solution, add compound e (the molar ratio of compound e to compound d is 0.9:1), stir, and add the resulting solution to the diazonium salt solution, keeping the temperature not exceeding 8°C. Add the above-prepared sodium hydroxide aqueous solution dropwise, adjust the pH to 8-10, and keep the temperature at 5-10°C for 3-4h. Add hydrochloric acid solution dropwise to adjust the pH to 2-3, keep the temperature not exceeding 25°C, filter, wash the filter cake with purified water until the filtrate is neutral, filter again, and collect the filter cake. Pump 48.0kg of tetrahydrofuran aqueous solution (22.5kg tetrahydrofuran, 25.5L purified water) into the reactor, add the above-obtained filter cake, beat, filter, wash the filter cake with tetrahydrofuran aqueous solution, wash the filter cake with purified water, filter again, and collect the filter cake. Dry the filter cake. |
| Ethyl acetate was pumped into the reactor, and the above-obtained materials were added to the reactor for slurrying, and the filter cake was washed with ethyl acetate, and the filter cake was washed until no obvious droplets flowed out of the mirror, and the filter cake was collected and dried to obtain the compound of formula (I-2). The yield in this step was 5.34 kg, and the yield was 97.5%. |
| Example 4. Synthesis of (Z)-5-(2-hydroxy-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazol-4(5H)-ylidene)hydrazino)phenyl)furan-2-carboxylic acid |
| The compound of formula (I-2) was prepared by using a method substantially the same as in Example 3 (except that the equivalent of compound e was adjusted from 0.9 in Example 3 to the current 0.95, other conditions remained unchanged). |
| Comparative Example 1: Synthesis of (Z)-5-(2-hydroxy-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazole-4(5H)-ylidene)hydrazino)phenyl)furan-2-carboxylic acid |
| |
| The compound of formula (I-2) was prepared by using a method substantially the same as in Example 3 (except that the step of adding urea was changed to starch potassium iodide test paper to indicate the reaction endpoint, and other conditions remained unchanged). |
| Test Example 1: Effect of urea on the preparation process of the compound of formula (I-2) |
| HPLC conditions: |
| Chromatographic column: Welch Ultimate |
| Flow rate: 1.0ml/min |
| Injection volume: 10 μl |
| Detector: UV detector |
| Detection wavelength: 251nm |
| Mobile phase: 0.1% trifluoroacetic acid aqueous solution was used as mobile phase A, acetonitrile was used as mobile phase B, and elution was performed at a ratio of 50%/50% of mobile phase A/mobile phase B. |
PATENT
EP 2441457
PATENT
WO 2010142137
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010142137&_cid=P21-MDCUXF-51461-1
PATENT
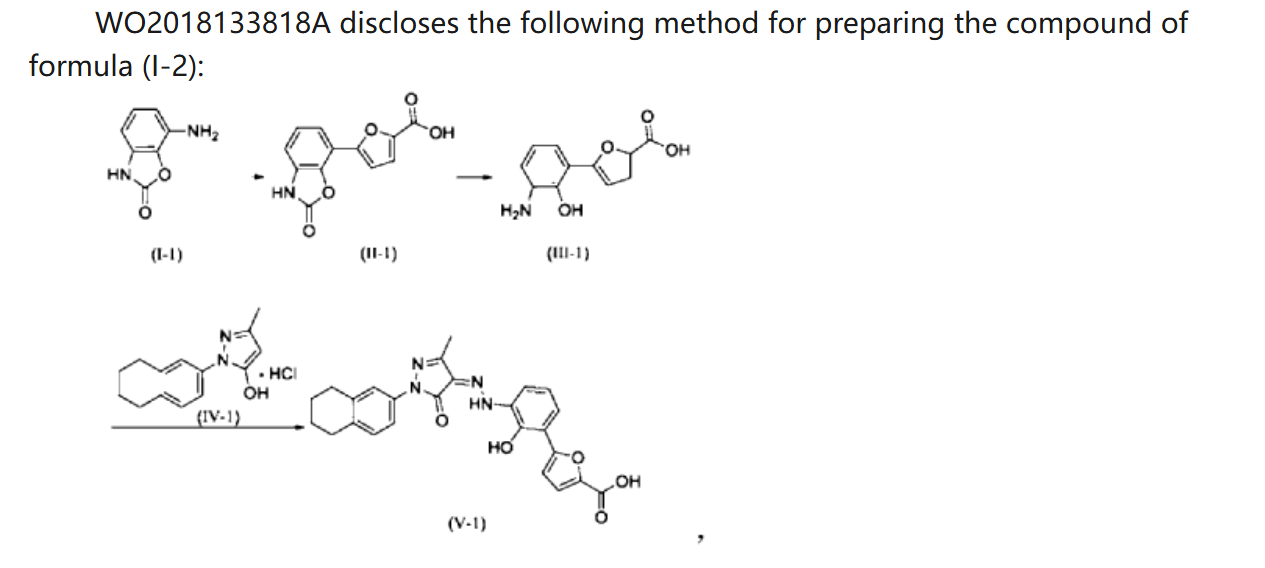
WO 2018133818
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018133818&_cid=P21-MDCUYN-53075-1
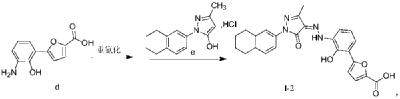
Example 1. Preparation of 3-methyl-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazol-5-ol hydrochloride
[0107]
[0108](5,6,7,8-tetrahydronaphthalene-2-yl)hydrazine hydrochloride (1.3 kg, prepared according to the method in patent application WO2009092276A1) and ethyl acetoacetate (1.17 L) were added to ethyl acetate (5.2 L). The mixture was heated under reflux for 2 hours. The reaction solution was cooled to room temperature, then cooled to 0-5°C, stirred for 1 hour, filtered, and the solid was washed with a small amount of ethyl acetate to obtain a white solid product (1.4 kg, yield 81%).
[0109]
[0110]Example 2. Preparation of (Z)-5-(2-hydroxy-3-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1,5-dihydro-4H-pyrazol-4-ylidene)hydrazino)phenyl)furan-2-carboxylic acid (V-1)
[0111]
[0112]Step 1: Synthesis of intermediate (II-1)
[0113]Purified water (14.80 kg), 7-aminobenzo[d]oxazol-2(3H)-one (2.00 kg, prepared according to the method in patent application WO2005016898A2), and hydrochloric acid (5.33 kg) were added to the reaction kettle, the temperature was raised to 40-45°C, stirred for 10 min, cooled to -3-5°C, and sodium nitrite aqueous solution (sodium nitrite 940 g, water 3.20 kg) was added dropwise, the internal temperature was kept at no more than 5°C, the end point was controlled by starch potassium iodide test paper, and stirring was continued for 15 min;
[0114]Add acetone (28L) to the reactor, then add furoic acid (4.57kg) and cupric chloride dihydrate (232g), stir at 35-40℃ until dissolved, add diazonium salt solution dropwise, keep the internal temperature at 35-40℃, and continue stirring for 1.5h. Add purified water (60L), heat to 35-40℃ and stir for 30min. Filter, wash the filter cake with 45-50℃ purified water. Add the filter cake to purified water (40kg), adjust the pH to 8-9 with 15% sodium hydroxide aqueous solution, filter, adjust the pH of the filtrate to 3-4 with 6mol/L hydrochloric acid, filter, wash the filter cake with purified water, and dry to obtain a solid (1.63kg, yield 50%).
[0115]Step 2: Synthesis of intermediate (III-1)
[0116]The product from the previous step (1.4 kg) and 15% aqueous sodium hydroxide solution (9.7 kg) were heated to reflux under argon protection and reacted for 28 hours. The reaction solution was poured into ice water (5-6 kg), and hydrochloric acid (6N, 3 L) was slowly added to adjust the pH value to 5-6. The temperature was maintained below 20°C. During this period, ethyl acetate was added to eliminate bubbles. The mixture was filtered, washed with purified water, and dried to obtain a solid (1.18 kg, yield 94%).
[0117]Step 3: Synthesis of intermediate (V-1)
[0118]Add the product of the previous step (1.10kg), purified water (27.5kg), and hydrochloric acid (2.92kg) to the reactor in sequence, stir and dissolve, cool to -4 to -1°C, add sodium nitrite aqueous solution (346g sodium nitrite, 5.5kg water), and continue to react for 15min after the addition is completed. Cool to -8 to -5°C. Dissolve sodium hydroxide (1.48kg) in purified water (13.2kg) to obtain a 10% sodium hydroxide aqueous solution. Add 5-methyl-2-(5,6,7,8-tetrahydronaphthalen-2-yl)-2H-pyrazole-3-ol hydrochloride (1.26kg) to the above sodium hydroxide aqueous solution (10kg) to dissolve, and add the resulting solution to the diazonium salt solution at once, keeping the temperature not higher than 10°C. Add the remaining 10% sodium hydroxide aqueous solution, adjust the pH to 8 to 9, naturally heat to 8 to 12°C for reaction, and react for 4h. Add 6N hydrochloric acid, adjust pH=2-3, keep the temperature not more than 20°C, filter, and wash the filter cake with water until pH=6-7. Add the filter cake to 50% tetrahydrofuran aqueous solution (19kg), slurry at room temperature for 2h, filter, wash with 50% tetrahydrofuran aqueous solution, wash with water, and dry. Add ethyl acetate (20kg) to the solid, slurry at 40-45°C for 2h under argon protection, cool to room temperature, filter, wash with ethyl acetate, add the solid to ethyl acetate (20kg), slurry at 40-45°C for 2h under argon protection, cool to room temperature, filter, wash with ethyl acetate, and dry to obtain a solid (2.18kg, yield 95%, purity 99.5%).
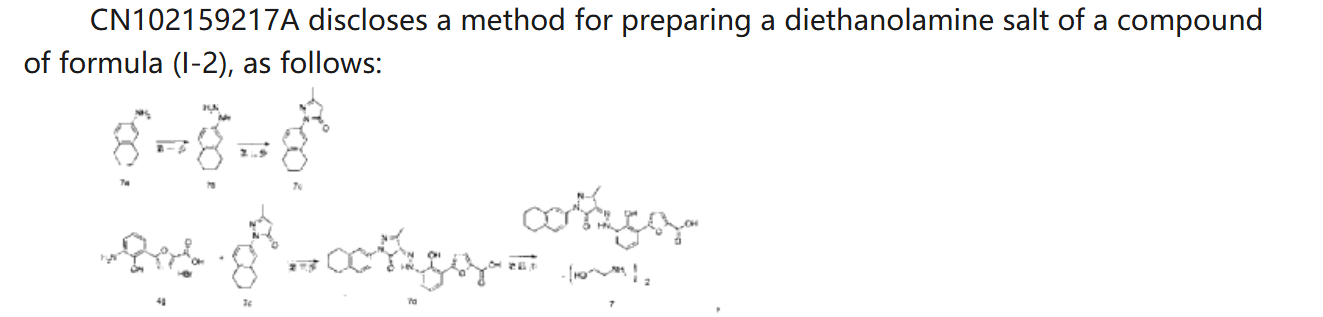
[0120]Example 3. Preparation of (Z)-5-(2-hydroxy-3-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1,5-dihydro-4H-pyrazol-4-ylidene)hydrazino)phenyl)furan-2-carboxylic acid ethanolamine salt (1:2)
[0121]
[0122]Preparation of crude product
[0123]The compound of formula (V-1) (1.8 kg) was suspended in a tetrahydrofuran/ethanol (14.5 kg, V/V = 2:1) mixed solvent at room temperature, stirred for 0.5 h, cooled to 10-15 ° C, and a tetrahydrofuran ethanol solution of ethanolamine (479.6 g) (tetrahydrofuran 91 g and ethanol 41 g) was added dropwise. The mixture was naturally heated to room temperature and reacted for 20 h. Filtered, washed with a tetrahydrofuran/ethanol (V/V = 2:1) mixed solvent, washed with ethyl acetate, filtered, and dried to obtain a dark red solid (1.73 kg, yield 76%, purity 99.7%).
[0124]
1H-NMR(500MHz,D 2O+NaOH)δ7.725-7.741(d,1H),7.298-7.316(d,3H),7.183-7.198(d,1H),7.131-7.149(m,2H),6.612-6.643(t,1H),3.574-3.596(t,4H),2.759-2.778(br,4H),2.698-2.721(t,4H),2.428(s,3H),1.772(br,4H).
SYN
J.Med.Chem.2024,67,4376−4418
HetrombopagOlamine (Hengqu).
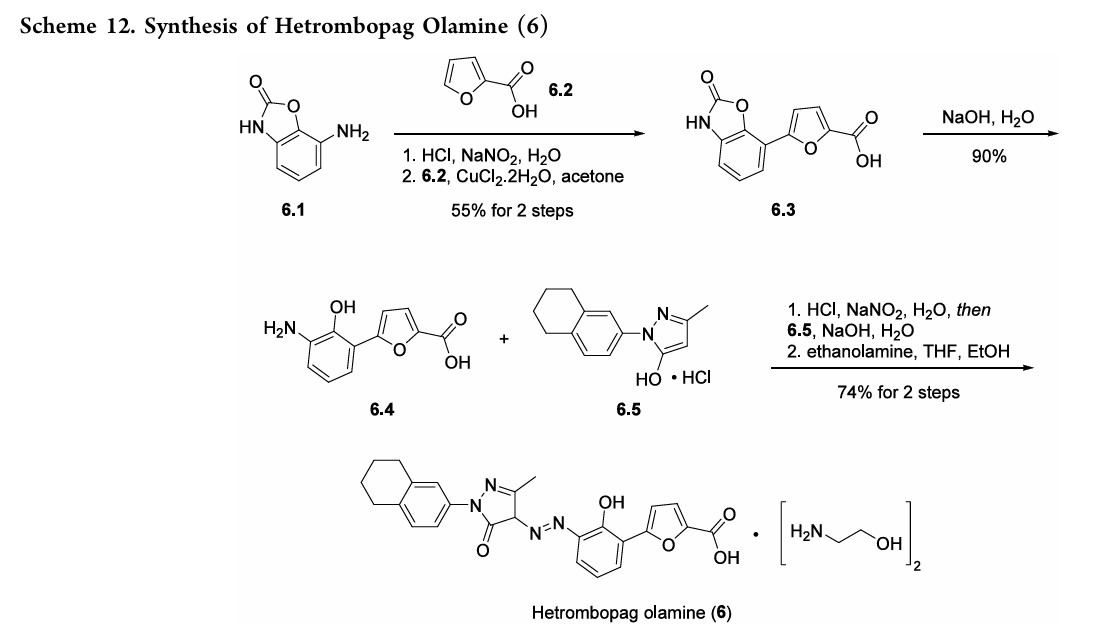
Hetrombopag olamine (6), an oral nonpeptide thrombopoietin receptor
(TpoR)agonistdevelopedby JiangsuHengruiPharmaceutical, was approved in China in June2021 for treatment of adult patients with chronic primary immune thrombocytopenia (ITP) and severe aplastic anemiawhohave not responded well to other treatments.46Hetrombopag, like other TpoR agonists, increases platelet production by binding to the transmembranedomainofTpoRinprogenitorcells, inducing
megakaryocytes.Theeffectisadditivewiththeactionofnative thrombopoietin, whichbinds to the extracellular domainof TpoR.Hetrombopag is structurallyrelatedtoeltrombopag, a previously approvedTpoR, withmodifications to enhance potencyandminimizetoxicity.46−48InaPhaseIIIclinicaltrial, ITPpatients demonstratedadurableplatelet response, with reducedbleedingriskanduseof rescuetherapycomparedto
placebo.49 Akilo-scale, chromatography-freesynthesisofhetrombopag has been reported by researchers at Jiangsu Hengrui Pharmaceutical in the Chinese-language patent literature (Scheme 12).50,51 Commercially available aniline 6.1 was coupledwith furoic acid (6.2) using aMeerwein arylation reaction togive intermediate6.3.This process first involves diazotizationof the anilineusing sodiumnitrite andhydrochloricacid.Ureawasusedtoquenchtheresidualnitrousacid, animprovement thatultimatelygavetheproductwithhigher purity and lower levels of specific impurities; the crude
diazoniumsalt solutionwas carried forwarddirectlywithout furthermanipulation.Furoicacid(6.2)inacetonewastreated withcopper(II)chloridedihydratefollowedbyadditionofthe
diazonium salt solution to affect the arylation. The crude productwaspurifiedbyacid−baseextractionandisolatedby filtrationtoprovide6.3 in55%yield.Basichydrolysisof the
cycliccarbamateunveiledthefreeanilineandphenolmoieties in arene 6.4. Nucleophilic attack of the enolate anion of pyrazolone 6.5 (see Scheme 13) on the diazoniumsalt of aniline6.4 formed the central hydrazonemoiety ina JappKlingemann-like reaction. The crude product was triturated withethylacetatetorapidlyprovidehetrombopagfreebasein
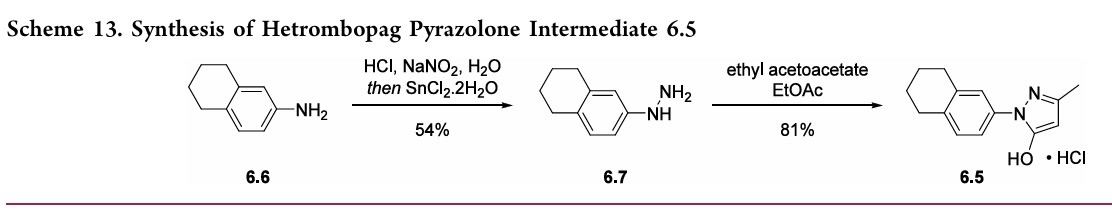
97.5%yield.TreatmentwithethanolamineinTHFandEtOH thengeneratedhetrombopagolamine (6) in76%yieldand 99.7%purity.51 Pyrazolone intermediate6.5was synthesized in two steps
(Scheme 13).52,53 5,6,7,8-Tetrahydronaphthalen-2-yl amine (6.6)was converted to the diazoniumion and reduced in situ to the corresponding hydrazine 6.7 using stannous chloridedihydrate.Condensationof thehydrazinewithethyl acetoacetate in ethyl acetate and in situ cyclization gave pyrazolone6.5.While the synthesis fromaniline6.1 to the activepharmaceutical ingredient(API)6wasreportedonthe
kilo-scale, thesynthesisofpyrazolone6.5wasreportedonlyon gram-scale
(46) Syed, Y. Y. Hetrombopag: First approval. Drugs 2021, 81, 1581−1585.
(47) Xie, C.; Zhao, H.; Bao, X.; Fu, H.; Lou, L. Pharmacological characterization of hetrombopag, a novel orally active human thrombopoietin receptor agonist. J. Cell. Mol. Med. 2018, 22, 5367−5377.
(48) Zheng, L.; Liang, M.-z.; Zeng, X.-l.; Li, C.-z.; Zhang, Y.-f.; Chen, X.-y.; Zhu, X.; Xiang, A.-b. Safety, pharmacokinetics and pharmacodynamics of hetrombopag olamine, a novel TPO-R agonist, in healthy individuals. Basic Clin. Pharmacol. Toxicol. 2017, 121, 414−422.
(49) Mei, H.; Liu, X.; Li, Y.; Zhou, H.; Feng, Y.; Gao, G.; Cheng, P.; Huang, R.; Yang, L.; Hu, J.; Hou, M.; Yao, Y.; Liu, L.; Wang, Y.; Wu, D.; Zhang, L.; Zheng, C.; Shen, X.; Hu, Q.; Liu, J.; Jin, J.; Luo, J.; Zeng, Y.; Gao, S.; Zhang, X.; Zhou, X.; Shi, Q.; Xia, R.; Xie, X.; Jiang, Z.; Gao, L.; Bai, Y.; Li, Y.; Xiong, J.; Li, R.; Zou, J.; Niu, T.; Yang, R.;
Hu, Y. A multicenter, randomized phase III trial of hetrombopag: a novel thrombopoietin receptor agonist for the treatment of immune thrombocytopenia. J. Hematol. Oncol. 2021, 14, 37.
(50) Shi, A.; Diao, A.; Du, Y. Preparation of bicyclic substituted pyrazolone azo derivatives. China Patent CN 113929668, 2022.
(51) Diao, A.; Gao, X.; Bian, L. Method for preparing bicyclo substituted pyrazolone azo derivatives and intermediates. WO 2018133818, 2018.
(52) Tang, P. C.; Lue, H.; Fei, H.; Chen, Y. Preparation of pyrazole derivatives as thrombopoietin receptor agonists. WO 2010142137, 2010.
(53) Tang, P. C.; Lue, H.; Fei, H.; Chen, Y. Salts of bicyclo substituted pyrazolon azo derivatives, preparation method and use
thereof. European Patent EP 2441457, 2014.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
//////////Hetrombopag Olamine, CHINA 2021, APPROVALS 2021, Hetrombopag diolamine, SHR 8735 olamine, Hetrombopag ethanolamine, SHR-8735 olamine, V45T2I862X, RAFUTROMBOPAG OLAMINE