















IMETELSTAT
CAS 868169-64-6, N163L
Molecular Formula, C148-H211-N68-O53-P13-S13, Molecular Weight, 4610.2379,
Nucleic Acid Sequence
Sequence Length: 135 a 1 c 4 g 3 tmodified
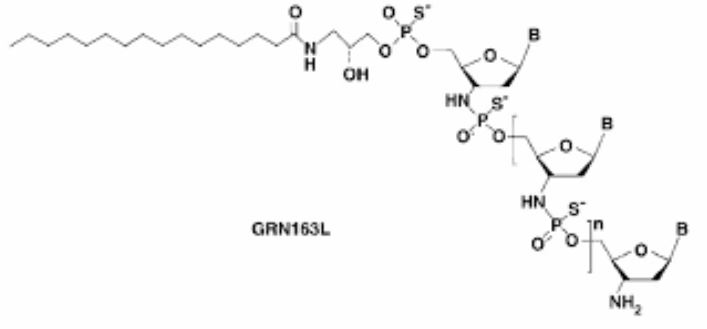
DNA d(3′-amino-3′-deoxy-P-thio)(T-A-G-G-G-T-T-A-G-A-C-A-A) 5′-[O-[2-hydroxy-3-[(1-oxohexadecyl)amino]propyl] hydrogen phosphorothioate]
PHASE 3, GERON, Myelodysplasia
IMETELSTAT SODIUM
CAS 1007380-31-5, GRN163L, GRN 163L Sodium Salt
| Molecular Formula: | C148H198N68Na13O53P13S13 |
|---|---|
| Molecular Weight: | 4895.941 g/mol |
5′-(O-(2-hydroxy-3-((1-oxohexadecyl)amino)propyl)phosphorothioate)-d(3′-amino-3′-deoxy-p-thio)(t-a-g-g-g-t-t-a-g-a-c-a-a), sodium salt (13)
DNA, d(3′-amino-3′-deoxy-p-thio)(T-A-G-G-G-T-T-A-G-A-C-A-A), 5′-(o-(2-hydroxy-3-((1-oxohexadecyl)amino)propyl) hydrogen phosphorothioate), sodium salt (1:13)
UNII-2AW48LAZ4I, Antineoplastic
In 2014, Geron entered into an exclusive worldwide license and collaboration agreement with Janssen Biotech for the treatment of hematologic cancers. However, in 2018, the agreement was terminated and Geron regained global rights to the product.
In 2015, imetelstat was granted orphan drug status in the U.S. for the treatment of myelodysplastic syndrome, as well as in both the U.S. and the E.U. for the treatment of myelofibrosis. In 2017, fast track designation was received in the U.S. for the treatment of adult patients with transfusion-dependent anemia due to low or intermediate-1 risk myelodysplastic syndromes (MDS) who are non-del(5q) and who are refractory or resistant to treatment with an erythropoiesis stimulating agent (ESA).
Imetelstat Sodium is the sodium salt of imetelstat, a synthetic lipid-conjugated, 13-mer oligonucleotide N3′ P5′-thio-phosphoramidate with potential antineoplastic activity. Complementary to the template region of telomerase RNA (hTR), imetelstat acts as a competitive enzyme inhibitor that binds and blocks the active site of the enzyme (a telomerase template antagonist), a mechanism of action which differs from that for the antisense oligonucleotide-mediated inhibition of telomerase activity through telomerase mRNA binding. Inhibition of telomerase activity in tumor cells by imetelstat results in telomere shortening, which leads to cell cycle arrest or apoptosis.
Imetelstat sodium, a lipid-based conjugate of Geron’s first-generation anticancer drug, GRN-163, is in phase III clinical trials at Geron for the treatment of myelodysplastic syndrome, as well as in phase II for the treatment of myelofibrosis. 
Geron is developing imetelstat, a lipid-conjugated 13-mer thiophosphoramidate oligonucleotide and the lead in a series of telomerase inhibitors, for treating hematological malignancies, primarily myelofibrosis.
Imetelstat, a first-in-class telomerase inhibitor and our sole product candidate, is being developed for the potential treatment of hematologic myeloid malignancies. Imetelstat is currently in two clinical trials being conducted by Janssen under the terms of an exclusive worldwide collaboration and license agreement.
Originally known as GRN163L, imetelstat sodium (imetelstat) is a 13-mer N3’—P5’ thio-phosphoramidate (NPS) oligonucleotide that has a covalently bound 5’ palmitoyl (C16) lipid group. The proprietary nucleic acid backbone provides resistance to the effect of cellular nucleases, thus conferring improved stability in plasma and tissues, as well as significantly improved binding affinity to its target. The lipid group enhances cell permeability to increase potency and improve pharmacokinetic and pharmacodynamic properties. The compound has a long residence time in bone marrow, spleen and liver. Imetelstat binds with high affinity to the template region of the RNA component of telomerase, resulting in direct, competitive inhibition of telomerase enzymatic activity, rather than elicit its effect through an antisense inhibition of protein translation. Imetelstat is administered by intravenous infusion.
Preclinical Studies with Imetelstat
A series of preclinical efficacy studies of imetelstat have been conducted by Geron scientists and academic collaborators. These data showed that imetelstat:
- Inhibits telomerase activity, and can shorten telomeres.
- Inhibits the proliferation of a wide variety of tumor types, including solid and hematologic, in cell culture systems and rodent xenograft models of human cancers, impacting the growth of primary tumors and reducing metastases.
- Inhibits the proliferation of malignant progenitor cells from hematologic cancers, such as multiple myeloma, myeloproliferative neoplasms and acute myelogenous leukemia.
- Has additive or synergistic anti-tumor effect in a variety of cell culture systems and xenograft models when administered in combination with approved anti-cancer therapies, including radiation, conventional chemotherapies and targeted agents.
Clinical Experience with Imetelstat
Over 500 patients have been enrolled and treated in imetelstat clinical trials.
PHASE 1
Six clinical trials evaluated the safety, tolerability, pharmacokinetics and pharmacodynamics both as a single agent and in combination with standard therapies in patients with solid tumors and hematologic malignancies:
- Single agent studies of imetelstat were in patients with advanced solid tumors, multiple myeloma and chronic lymphoproliferative diseases. Combination studies with imetelstat were with bortezomib in patients with relapsed or refractory multiple myeloma, with paclitaxel and bevacizumab in patients with metastatic breast cancer, and with carboplatin and paclitaxel in patients with advanced non-small cell lung cancer (NSCLC).
- Doses ranging from 0.5 mg/kg to 11.7 mg/kg were tested in a variety of dosing schedules ranging from weekly to once every 28 days.
- The human pharmacokinetic profile was characterized in clinical trials of patients with solid tumors and chronic lymphoproliferative diseases. Single-dose kinetics showed dose-dependent increases in exposure with a plasma half-life (t1/2) ranging from 4-5 hours. Residence time in bone marrow is long (0.19-0.51 µM observed at 41-45 hours post 7.5 mg/kg dose).
- Telomerase inhibition was observed in various tissues where the enzymes’s activity was measurable.
PHASE 2
Imetelstat was studied in two randomized clinical trials, two single arm proof-of-concept studies and an investigator sponsored pilot study:
- Randomized trials were in combination with paclitaxel in patients with metastatic breast cancer and as maintenance treatment following a platinum-containing chemotherapy regimen in patients with NSCLC.
- Single arm studies were as a single agent or in combination with lenalidomide in patients with multiple myeloma and as a single agent in essential thrombocythemia (ET) or polycythemia vera (PV).
- An investigator sponsored pilot study was as a single agent in patients with myelofibrosis (MF) or myelodysplastic syndromes (MDS).
SAFETY AND TOLERABILITY
The safety profile of imetelstat across the Phase 1 and 2 trials has been generally consistent. Reported adverse events (AEs) and laboratory investigations associated with imetelstat administration included cytopenias, transient prolonged activated partial thromboplastin time (aPTT; assessed only in Phase 1 trials), gastrointestinal symptoms, constitutional symptoms, hepatic biochemistry abnormalities, and infusion reactions. Dose limiting toxicities include thrombocytopenia and neutropenia.
A Focus on Hematologic Myeloid Malignancies
Early clinical data from the Phase 2 clinical trial in ET and the investigator sponsored pilot study in MF suggest imetelstat may have disease-modifying activity by suppressing the proliferation of malignant progenitor cell clones for the underlying diseases, and potentially allowing recovery of normal hematopoiesis in patients with hematologic myeloid malignancies.
Results from these trials were published in the New England Journal of Medicine:
- Baerlocher GM, et al. Telomerase Inhibitor Imetelstat in Patients with Essential Thrombocythemia. N Engl J Med. 2015 Sep 3; 373(10):920-8
- Tefferi A, et al. A Pilot Study of the Telomerase Inhibitor Imetelstat for Myelofibrosis. N Engl J Med. 2015 Sep 3; 373(10):908-19
Current Clinical Trials
Imetelstat is currently being tested in two clinical trials: IMbark, a Phase 2 trial in myelofibrosis (MF), and IMerge, a Phase 2/3 trial in myelodysplastic syndromes (MDS).
IMbark
IMbark is the ongoing Phase 2 clinical trial to evaluate two doses of imetelstat in intermediate-2 or high-risk MF patients who are refractory to or have relapsed after treatment with a JAK inhibitor.
Internal data reviews were completed in September 2016, April 2017 and March 2018. The safety profile was consistent with prior clinical trials of imetelstat in hematologic malignancies, and no new safety signals were identified. The data supported 9.4 mg/kg as an appropriate starting dose in the trial, but an insufficient number of patients met the protocol defined interim efficacy criteria and new patient enrollment was suspended in October 2016. As of January 2018, median follow up was approximately 19 months, and median overall survival had not been reached in either dosing arm. In March 2018, the trial was closed to new patient enrollment. Patients who remain in the treatment phase of the trial may continue to receive imetelstat, and until the protocol-specified primary analysis, all safety and efficacy assessments are being conducted as planned in the protocol, including following patients, to the extent possible, until death, to enable an assessment of overall survival.
IMerge
IMerge is the ongoing two-part Phase 2/3 clinical trial of imetelstat in red blood cell (RBC) transfusion-dependent patients with lower risk MDS who are refractory or resistant to treatment with an erythropoiesis stimulating agent (ESA). Part 1 is a Phase 2, open-label, single-arm trial of imetelstat administered as a single agent by intravenous infusion, and is ongoing. Part 2 is designed to be a Phase 3, randomized, controlled trial, and has not been initiated.
Preliminary data as of October 2017 from the first 32 patients enrolled in the Part 1 (Phase 2) of IMerge were presented as a poster at the American Society of Hematology Annual Meeting in December 2017.
The data showed that among the subset of 13 patients who had not received prior treatment with either lenalidomide or a hypomethylating agent (HMA) and did not have a deletion 5q chromosomal abnormality (non-del(5q)), 54% achieved RBC transfusion-independence (TI) lasting at least 8 weeks, including 31% who achieved a 24-week RBC-TI. In the overall trial population, the rates of 8- and 24-week RBC-TI were 38% and 16%, respectively. Cytopenias, particularly neutropenia and thrombocytopenia, were the most frequently reported adverse events, which were predictable, manageable and reversible.
Based on the preliminary data from the 13-patient subset, Janssen expanded Part 1 of IMerge to enroll approximately 20 additional patients who were naïve to lenalidomide and HMA treatment and non-del(5q) to increase the experience and confirm the benefit-risk profile of imetelstat in this refined target patient population
PATENT
WO 2005023994
WO 2006113426
WO 2006113470
| WO 2006124904 |
WO 2008054711
WO 2008112129
US 2014155465
WO 2014088785
PATENT
WO 2016172346
PATENT
WO2018026646
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018026646
Patients of acute myeloid leukemia (AML) have limited treatment options at diagnosis; treatment typically takes the form of chemotherapy to quickly reduce the leukemic cell burden. Invasive leukapheresis procedures to remove large numbers of leukocytes (normal and diseased) may be applied in parallel to chemotherapy to temporarily lower tumor cell burden. Induction phase chemotherapy can be successful but, most healthy cells residing in patient bone marrow are also killed, causing illness and requiring additional palliative therapy to ward off infection and raise leukocyte counts. Additional rounds of chemotherapy can be used in an attempt to keep patients in remission; but relapse is common.
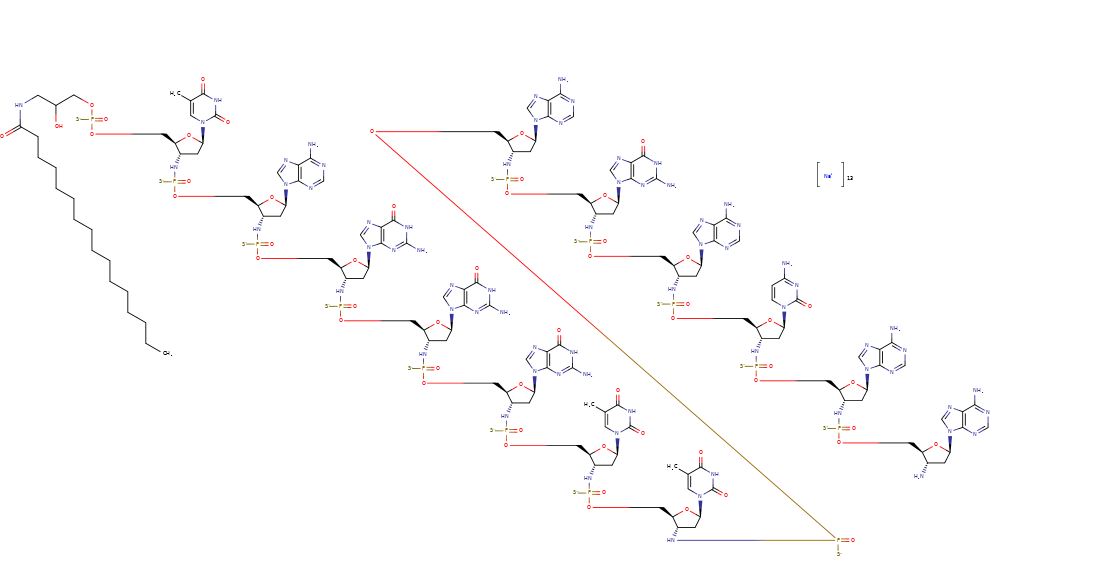
[0005] Telomerase is present in over 90% of tumors across all cancer types; and is lacking in normal, healthy tissues. Imetelstat sodium is a novel, first-in-class telomerase inhibitor that is a covalently-lipidated 13-mer oligonucleotide (shown below) complimentary to the human telomerase RNA (hTR) template region. Imetelstat sodium does not function through an anti-sense mechanism and therefore lacks the side effects commonly observed with such therapies. Imetelstat sodium is the sodium salt of imetelstat (shown below):
Imetelstat sodium
Unless otherwise indicated or clear from the context, references below to imetelstat also include salts thereof. As mentioned above, imetelstat sodium in particular is the sodium salt of imetelstat.
[0006] ABT-199/venetoclax (trade name Venclexta) is an FDA approved Bcl-2 inhibitor for use in chronic lymphocytic leukemia (CLL) patients with dell7p who are relapsed/refractory. ABT-199 is also known as ABT 199, GDC0199, GDC-0199 or RG7601. The chemical name for ABT-199 is 4-[4-[[2-(4-chlorophenyl)-4,4-dimethylcyclohexen-l-yl]methyl]piperazin-l-yl]-N-[3-nitro-4-(oxan-4-ylmethylamino)phenyl]sulfonyl-2-(lH-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide (Cas No. 1257044-40-8). Unless otherwise indicated or clear from the context, references below to ABT-199 also include pharmaceutically acceptable salts thereof. Specifically in the Examples however, ABT-199 was used in the free base form.
[0007] ABT-199, shown below in the free base form, is highly specific to Bcl-2, unlike other first generation inhibitors which show affinity for related Bel family members and induce greater side effects. Inhibition of Bcl-2 blocks the pro-apoptotic signals caused by damage to or abnormalities within cellular DNA and ultimately leads to programmed cell death in treated cells via the caspase cascade and apoptosis through the intrinsic pathway.
ABT-199 (shown in the free base form)
PATENT
WO-2019011829
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019011829&tab=PCTDESCRIPTION&maxRec=1000
Improved process for preparing imetelstat . claiming use of a combination comprising a telomerase inhibitor, specifically imetelstat sodium and a Bcl-2 inhibitor, specifically ABT-199 for treating hematological cancer such as acute myeloid leukemia, essential thrombocythemia and polycythemia vera, specifically acute myeloid leukemia.
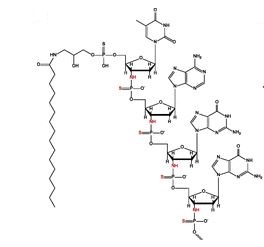
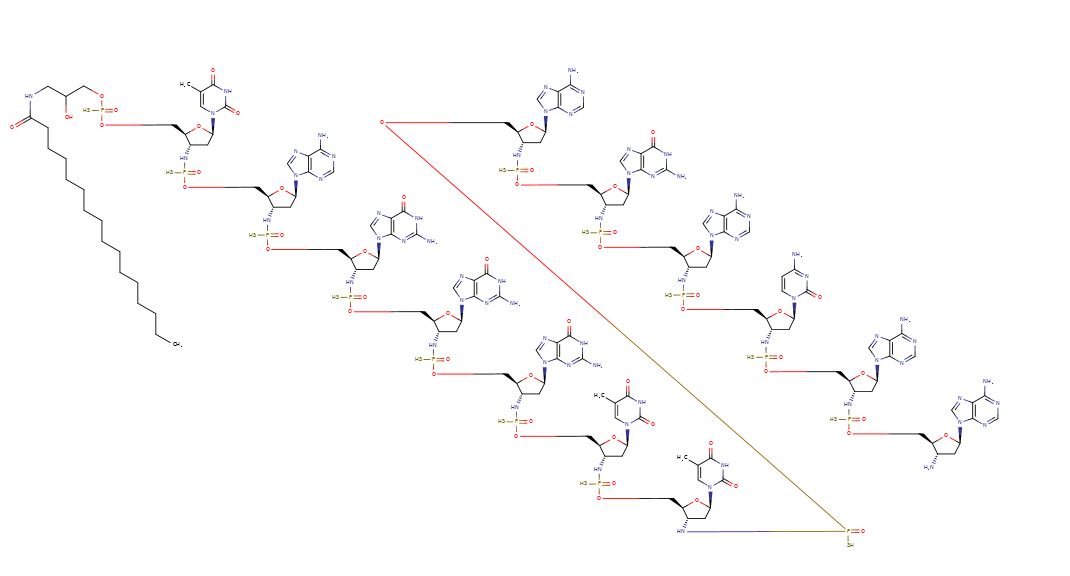
Imetelstat (SEQ ID NO: 1 ) is a N3′- P5′ thiophosphoramidate oligonucleotide covalently linked to a palmitoyl lipid moiety and has been described in WO-2005/023994 as compound (1 F). The sodium salt of imetelstat acts as a potent and specific telomerase inhibitor and can be used to treat telomerase-mediated disorders, e.g. cancer, including disorders such as myelofibrosis (MF), myelodysplastic syndromes (MDS) and acute myelogenous leukemia (AML).
The structure of imetelstat sodium is shown below :
The structure of imetelstat can also be represented as shown below
imetelstat
The LPT group represents the palmitoyi lipid that is covalently linked to the N3′- P5′ thiophosphor-amidate oligonucleotide. The base sequence of the thirteen nucleotides is as follows :
TAGGGTTAGACAA and is represented by the bases B1 to B13. The -NH-P(=S)(OH)-and -0-P(=S)(OH)- groups of the structure can occur in a salt form. It is understood that salt forms of a subject compound are encompassed by the structures depicted herein, even if not specifically indicated.
Imetelstat sodium can also be represented as follows
o H
LPT = CH3-(CH2)i4-C-N-CH2-(CHOH)-CH2-
The -NH-P(=S)(OH)- group and the thymine, adenine, guanine and cytosine bases can occur in other tautomeric arrangements then used in the figures of the description. It is understood that all tautomeric forms of a subject compound are encompassed by a structure where one possible tautomeric form of the compound is described, even if not specifically indicated.
Prior art
The synthetic scheme used in WO-2005/023994 to prepare imetelstat as compound (1 F) is described in Scheme 1 and Scheme 2. The synthesis of this oligonucleotide is achieved using the solid-phase phosphoramidite methodology with all reactions taking place on solid-phase support. The synthesis of imetelstat is carried out on controlled pore glass (LCAA-CPG) loaded with
3-palmitoylamido-1-0-(4, 4′-dimethoxytrityl)-2-0-succinyl propanediol. The oligonucleotide is assembled from the 5′ to the 3′ terminus by the addition of protected nucleoside 5′-phosphor-amidites with the assistance of an activator. Each elongation cycle consists of 4 distinct, highly controlled steps : deprotection, amidite coupling, sulfurization and a capping step.
Scheme 1 : imetelstat synthetic scheme cycle 1
3. Sulfurization
In Scheme 1 the solid-phase supported synthesis starts with removal of the acid-labile 4,4-dimethoxy-trityl (DMT) protecting group from the palmitoylamidopropanediol linked to the solid-phase support. The first phosphoramidite nucleotide is coupled to the support followed by sulfurization of the phosphor using a 0.1 M solution of phenylacetyl disulfide (PADS) in a mixture of acetonitrile and 2,6-lutidine (1 : 1 ratio). Then a capping step is applied to prevent any unreacted solid-phase support starting material from coupling with a phosphoramidite nucleotide in the following reaction cycles. Capping is done using an 18:1 :1 mixture of THF / isobutyric anhydride / 2,6-lutidine.
After the first cycle on the solid-phase support, chain elongation is achieved by reaction of the 3′-amino group of the support-bound oligonucleotide with an excess of a solution of the protected nucleotide phosphoramidite monomer corresponding to the next required nucleotide in the sequence as depicted in Scheme 2.
Scheme 2 : imetelstat synthetic scheme cycle 2-13
In Scheme 2 the first cycle is depicted of the chain elongation process which is achieved by deprotection of the 3′-amino group of the support-bound oligonucleotide (a), followed by a coupling reaction of the 3′-amino group of the support-bound oligonucleotide (b) with an excess of a solution of a 5′-phosphoramidite monomer corresponding to the next required nucleotide in the sequence of imetelstat. The coupling reaction is followed by sulfurization of the phosphor of the support-bound oligonucleotide (c) and a capping step (see Scheme 3) to prevent any unreacted solid-phase support starting material (b) from coupling with a 5′-phosphoramidite nucleotide in the following reaction cycles. The reaction cycle of Scheme 2 is repeated 12 times before the solid-phase support-bound oligonucleotide is treated with a 1 :1 mixture of ethanol and concentrated ammonia, followed by HPLC purification to obtain imetelstat.
Scheme 3
The capping step using an 18:1 : 1 mixture of THF / isobutyric anhydride / 2,6-lutidine is done to convert after the coupling step any remaining solid-phase support bound oligonucleotide (b) with a primary 3′-amino group into oligonucleotide (e) with a protected (or ‘capped’) 3′-amino group in order to prevent the primary 3′-amino group from coupling with a phosphoramidite nucleotide in the next reaction cycles.
WO-01/18015 discloses in Example 3 with SEQ ID No. 2 a N3’^P5′ thiophosphoramidate oligonucleotide and a process for preparing this oligonucleotide encompassing a capping step.
Herbert B-S et al. discusses the lipid modification of GRN163 (Oncogene (2005) 24, 5262-5268).
Makiko Horie et al. discusses the synthesis and properties of 2′-0,4′-C-ethylene-bridged nucleic acid oligonucleotides targeted to human telomerase RNA subunit (Nucleic Acids Symposium Series (2005) 49, 171-172).
Description of the invention
The coupling reaction in the solid-phase support bound process disclosed in WO-01/18015 and WO-2005/023994 include a capping step to prevent any unreacted primary 3′ amino groups on the support-bound oligonucleotide from reacting during subsequent cycles.
It has now surprisingly been found that the use of a capping step as described in the prior art is superfluous and that imetelstat can be prepared using a 3-step cycle without an additional capping step with nearly identical yield and purity compared to the prior art 4-step cycle that uses a specific capping step. Eliminating the capping step from each cycle benefits the overall process by reducing the number of cycle steps by 22% (from 54 to 42 steps) and consequent reduction of process time. Also, the solvent consumption is reduced due to the reduction of cycle steps which makes for a greener process.
Wherever the term “capping step” is used throughout this text, it is intended to define an additional chemical process step wherein the primary free 3′-amino group on the solid-phase support bound oligonucleotide is converted into a substituted secondary or tertiary 3′-amino group that is not capable of participating in the coupling reaction with a protected 3′-aminonucleoside-5′-0-cyanoethyl-N,N-diisopropylamino-phosphoramidite monomer in the ensuing coupling step.
In one embodiment, the present invention relates to a method of synthesizing an oligonucleotide N3′ – P5′ thiophosphoramidate of formula
imetelstat
the method comprises of
a) providing a first 3′-amino protected nucleotide attached to a solid-phase support of formula (A) wherein PG is an acid-labile protecting group;
b) deprotecting the protected 3′-amino group to form a free 3′-amino group;
c) reacting the free 3′-amino group with a protected 3′-aminonucleoside-5′-0-cyanoethyl-N,N- diisopropylaminophosphoramidite monomer of formula (B n) wherein n = 2 to form an internucleoside N3′- P5′-phosphoramidite linkage;
mer (B’n)
d) sulfurization of the internucleoside phosphoramidite group using an acyl disulfide to form a N3′- P5′ thiophosphoramidate;
e) repeating 1 1 times in successive order the deprotection step b), the coupling step c) with a protected 3′-aminonucleoside-5′-0-cyanoethyl-N,N-diisopropylamino-phosphoramidite monomer of formula (B n) wherein the protected nucleoside base B’ in monomer (B n) is successively the protected nucleobase B3 to B13 in the respective 1 1 coupling steps, and the sulfurization step d);
f) removing the acid-labile protecting group PG; and
g) cleaving and deprotecting imetelstat from the solid-phase support;
characterized in that no additional capping step is performed in any of the reaction steps a) to e).
In one embodiment, the present invention relates to a method of synthesizing the N3′ – P5′
thiophosphoramidate oligonucleotide imetelstat of formula
imetelstat
the method comprises of
a) providing a first 3′-amino protected nucleotide attached to a solid-phase support of formula (A) wherein PG is an acid-labile protecting group;
b) deprotecting the protected 3′-amino group to form a free 3′-amino group;
c) reacting the free 3′-amino group with a protected 3′-aminonucleoside-5′-0-cyanoethyl- Ν,Ν-diisopropylaminophosphoramidite monomer of formula (B n), wherein B n with n = 2 is protected A, to form an internucleoside N3′- P5′-phosphoramidite linkage;
mer
d) sulfurization of the internucleoside phosphoramidite group using an acyl disulfide to form a N3′- P5′ thiophosphoramidate;
e) repeating 1 1 times in successive order the deprotection step b), the coupling step c) with a protected 3′-aminonucleoside-5′-0-cyanoethyl-N,N-diisopropylamino-phosphoramidite monomer of formula (B n) wherein the nucleoside base B’ of monomer (B n) is protected B except when B is thymine, and wherein Bn is successively nucleobase B3 to B13 in the respective 1 1 coupling steps, and the sulfurization step d);
f) removing the acid-labile protecting group PG; and
g) deprotecting and cleaving imetelstat from the solid-phase support;
characterized in that no additional capping step is performed in any of the reaction steps a) to e).
In one embodiment, the present invention relates to a method of synthesizing the N3′ – P5′
thiophosphoramidate oligonucleotide imetelstat of formula
imetelstat
thymine
adenine
guanine
cytosine
9 H
LPT =CH3-(CH2)i4-C-N-CH2-(CHOH)-CH2-
the method comprises of
a) providing a first protected 3′-amino nucleotide attached to a solid-phase support of formula (A) wherein PG is an acid-labile protecting group;
b) deprotecting the PG-protected 3′-amino nucleotide to form a free 3′-amino nucleotide of formula (A’);
c) coupling the free 3′-amino nucleotide with a protected 3′-aminonucleoside-5′-0- cyanoethyl-N,N-diisopropylaminophosphoramidite monomer (B n), wherein B nwith n = 2 is protected A, to form an internucleoside N3′- P5′-phosphoramidite linkage;
monomer (B’n)
d) sulfurizing the N3′- P5′-phosphoramidite linkage using an acyl disulfide to form an internucleoside N3′- P5′ thiophosphoramidate linkage;
e) repeating 1 1 times in successive order:
the deprotecting step b);
the coupling step c) with a protected 3′-aminonucleoside-5′-0-cyanoethyl-N,N- diisopropylamino-phosphoramidite monomer (B n) wherein the nucleoside base B’ of monomer (B n) is protected B except when B is thymine, and wherein Bn is successively nucleobase B3 to B13 in the respective 1 1 coupling steps; and
the sulfurizing step d);
to produce a protected N3′ – P5′ thiophosphoramidate oligonucleotide imetelstat attached to the solid-phase support;
f) removing the 3′-terminal acid-labile protecting group PG from the protected N3′ – P5′ thiophosphoramidate oligonucleotide imetelstat; and
g) deprotecting and cleaving the protected N3′ – P5′ thiophosphoramidate oligonucleotide imetelstat from the solid-phase support to produce imetelstat;
characterized in that no additional capping step is performed in any of the reaction steps a) to e).
A wide variety of solid-phase supports may be used with the invention, including but not limited to, such as microparticles made of controlled pore glass (CPG), highly cross-linked polystyrene, hybrid controlled pore glass loaded with cross-linked polystyrene supports, acrylic copolymers, cellulose, nylon, dextran, latex, polyacrolein, and the like.
The 3′-amino protected nucleotide attached to a solid-phase support of formula (A)
can be prepared as disclosed in WO-2005/023994 wherein a controlled pore glass support loaded with 3-palmitoylamido-1-0-(4, 4′-dimethoxytrityl)-2-0-succinyl propanediol has been coupled with a protected 3′-aminonucleoside-5′-0-cyanoethyl-N,N-diisopropylaminophosphoramidite monomer of formula (B^ )
monomer (B’-| ) wherein B’-| = T
wherein PG is an acid-labile protecting group. Suitable acid-labile 3′-amino protecting groups PG are, but not limited to, e.g. triphenylmethyl (i.e. trityl or Tr), p-anisyldiphenylmethyl (i.e. mono-methoxytrityl or MMT), and di-p-anisylphenylmethyl (i.e. dimethoxytrityl or DMT).
The protected 3′-aminonucleoside-5′-0-cyanoethyl-N,N-diisopropylaminophosphoramidite monomers of formula (B n) have a 3′-amino protecting group PG which is an acid-labile group, such as triphenylmethyl (i.e. trityl or Tr), p-anisyldiphenylmethyl (i.e. monomethoxytrityl or MMT), or di-p-anisylphenylmethyl (i.e. dimethoxytrityl or DMT). Furthermore the nucleoside base B’ is protected with a base-labile protecting group (except for thymine).
ed A ed C ed A ed A
B’s = protected A G = guanine
B’g = protected G C = cytosine
The nucleotide monomers and B’2 to B’13 are used successively in the 13 coupling steps starting from the provision of a solid-phase support loaded with 3-palmitoylamido-1-0-(4, 4′-dimethoxytrityl)-2-0-succinyl propanediol and coupled to nucleotide monomer and the following cycle of 12 deprotection, coupling, and sulfurization reactions wherein the nucleotide monomers B’2 to B -I 3 are used.
The 3′-amino protecting group PG can be removed by treatment with an acidic solution such as e.g. dichloroacetic acid in dichloromethane or toluene.
The nucleoside base B’ in the protected 3′-aminonucleoside-5′-0-cyanoethyl-N,N-diisopropyl-aminophosphoramidite monomers of formula (B n) is protected with a base-labile protecting group which is removed in step g). Suitable base-labile protecting groups for the nucleoside base adenine, cytosine or guanine are e.g. acyl groups such as acetyl, benzoyl, isobutyryl, dimethyl-formamidinyl, or dibenzylformamidinyl. Under the reaction conditions used in oligonucleotide synthesis the thymine nucleoside base does not require protection. Such protected 3′- amino-nucleoside-5′-0-cyanoethyl-N,N-diisopropylaminophosphoramidite monomers of formula (B N) having a 3′-amino protected with an acid-labile group protecting group PG and a nucleoside base B’ protected with a base-labile protecting group are commercially available or can be prepared as described in WO-2006/014387.
The coupling step c) is performed by adding a solution of protected 3′-aminonucleoside-5′-0-cyanoethyl-N,N-diisopropylaminophosphoramidite monomer of formula (BN) and a solution of an activator (or a solution containing the phosphoramidite monomer (BN) and the activator) to the reaction vessel containing the free amino group of an (oligo)nucleotide covalently attached to a solid support. The mixture is then mixed by such methods as mechanically vortexing, sparging with an inert gas, etc. Alternately, the solution(s) of monomer and activator can be made to flow through a reaction vessel (or column) containing the solid-phase supported (oligo)nucleotide with a free 3′-amino group. The monomer and the activator either can be premixed, mixed in the valve-block of a suitable synthesizer, mixed in a pre-activation vessel and preequilibrated if desired, or they can be added separately to the reaction vessel.
Examples of activators for use in the invention are, but not limited to, tetrazole, 5-(ethylthio)-1 H-tetrazole, 5-(4-nitro-phenyl)tetrazole, 5-(2-thienyl)-1 H-tetrazole, triazole, pyridinium chloride, and the like. Suitable solvents are acetonitrile, tetrahydrofuran, dichloromethane, and the like. In practice acetonitrile is a commonly used solvent for oligonucleotide synthesis.
The sulfurization agent for use in step d) is an acyl disulfide dissolved in a solvent. Art know acyl disulfides are e.g. dibenzoyl disulphide, bis(phenylacetyl) disulfide (PADS), bis(4-methoxybenzoyl) disulphide, bis(4-methylbenzoyl) disulphide, bis(4-nitrobenzoyl) disulphide and bis(4-chlorobenzoyl) disulfide.
Phenylacetyl disulfide (PADS) is a commonly used agent for sulfurization reactions that it is best ‘aged’ in a basic solution to obtain optimal sulfurization activity (Scotson J.L. et al., Org. Biomol. Chem., vol. 14, 10840 – 10847, 2016). A suitable solvent for PADS is e.g. a mixture of a basic solvent such as e.g. 3-picoline or 2,6-lutidine with a co-solvent such as acetonitrile, toluene, 1-methyl-pyrrolidinone or tetrahydrofuran. The amount of the basic solvent to the amount of the co-solvent can be any ratio including a 1 :1 ratio. Depending upon the phosphite ester to be converted into its corresponding thiophospate, both ‘fresh’ and ‘aged’ PADS can be used however ‘aged’ PADS has been shown to improve the rate and efficiency of sulfurization. ‘Aged’ PADS solutions are freshly prepared PADS solutions that were maintained some time before usage in the sulfurization reaction. Aging times can vary from a few hours to 48 hours and the skilled person can determine the optimal aging time by analysing the sulfurization reaction for yield and purity.
For the preparation of imetelstat in accordance with the present invention, a PADS solution in a mixture of acetonitrile and 2,6-lutidine, preferably in a 1 :1 ratio, with an aging time of 4 to 14 hours is used. It has been found that when 2,6-lutidine is used, limiting the amount of 2,3,5-collidine (which is often found as an impurity in 2,6-lutidine) below 0.1 % improves the efficiency of sulfurization and less undesirable phosphor oxidation is observed.
In step g) imetelstat is deprotected and cleaved from the solid-phase support. Deprotection includes the removal of the β-cyanoethyl groups and the base-labile protecting groups on the nucleotide bases. This can be done by treatment with a basic solution such as a diethylamine (DEA) solution in acetonitrile, followed by treatment with aqueous ammonia dissolved in an alcohol such as ethanol.
The reaction steps a) to f) of the present invention are carried out in the temperature range of 10°C to 40°C. More preferably, these reactions are carried out at a controlled temperature ranging from 15°C to 30°C. In particular reaction step b) of the present invention is carried out in the temperature range of 15°C to 30°C; more in particular 17°C to 27°C. In particular reaction step d) of the present invention is carried out in the temperature range of 17°C to 25°C; more in particular 18°C to 22°C; even more in particular 19°C. The step g) wherein imetelstat is deprotected and cleaved from the solid-phase support is carried out at a temperature ranging from 30°C to 60°C. Depending upon the equipment and the specific reaction conditions used, the optimal reaction temperature for each step a) to g) within the above stated ranges can be determined by the skilled person.
After each step in the elongation cycle, the solid-phase support is rinsed with a solvent, for instance acetonitrile, in preparation for the next reaction.
After step g), crude imetelstat is obtained in its ammonium salt form which is then purified by a preparative reversed phase high performance liquid chromatography (RP-HPLC) by using either polymeric or silica based resins to get purified imetelstat in triethyl amine form. An excess of a sodium salt is added, and then the solution is desalted by diafiltration thereby yielding imetelstat sodium which is then lyophilized to remove water.
Experimental part
‘Room temperature’ or ‘ambient temperature’ typically is between 21-25 °C.
Experiment 1 (no capping step)
All the reagents and starting material solutions were prepared including 3% dichloroacetic acid (DCA) in toluene, 0.5 M 5-(ethylthio)-1 H-tetrazole in acetonitrile, 0.15 M of all 4 nucleotide monomers of formula (B n) in acetonitrile, 0.2 M phenyl acetyl disulfide (PADS) in a 1 :1 mixture of acetonitrile and 2,6-lutidine and 20% DEA (diethylamine) in acetonitrile.
The oligonucleotide synthesis was performed in the direction of 5′ to 3′ utilizing a repetitive synthesis cycle consisting of detritylation followed by coupling, and sulfurization performed at ambient temperature.
A column (diameter : 3.5 cm) was packed with a solid-support loaded with 3-palmitoylamido-1-0- (4, 4′-dimethoxytrityl)-2-0-succinyl propanediol (3.5 mmol based on a capacity of 400 μιηοΙ/g) that was coupled with the nucleotide monomer B Detritylation was achieved using 3% dichloroacetic acid (DCA) in toluene (amount is between 6.5 and 13.4 column volumes in each detritylation step) and the solid-support bound nucleotide was washed with acetonitrile (amount: 5 column volumes). Coupling with the next nucleotide monomer of formula (B n) was achieved by pumping a solution of 0.5 M 5-(ethylthio)-1 H-tetrazole in acetonitrile and 0.15 M of the next nucleotide monomer of formula (B n) in the sequence, dissolved in acetonitrile, through the column. The column was washed with acetonitrile (amount : 2 column volumes). Then sulfurization was performed by
pumping a solution of 0.2 M phenyl acetyl disulfide (PADS) in a 1 :1 mixture of acetonitrile and 2,6-lutidine mixture through the column followed by washing the column with acetonitrile (amount : 5 column volumes).
The synthesis cycle of detritylation, coupling with the next nucleotide monomer of formula (B n) and sulfurization was repeated 12 times, followed by detritylation using 3% dichloroacetic acid (DCA) in toluene (amount is between 6.5 and 13.4 column volumes).
Upon completion of the synthesis cycle, the crude oligonucleotide on the solid-support support was treated with a diethylamine (DEA) solution followed by treatment with ammonium hydroxide solution: ethanol (3: 1 volume ratio) at a temperature of 55°C. The reaction mixture was aged for
4 to 24 hours at 55°C, cooled to room temperature, and slurry was filtered to remove the polymeric support. The solution comprising imetelstat in its ammonium form was subjected to the HPLC analysis procedure of Experiment 3.
Experiment 2 (with capping step)
All the reagents and starting material solutions were prepared including 3% dichloroacetic acid (DCA) in toluene, 0.5 M 5-(ethylthio)-1 H-tetrazole in acetonitrile, 0.15 M of all 4 nucleotide monomers of formula (B n) in acetonitrile, 0.2 M phenyl acetyl disulfide (PADS) in a 1 :1 mixture of acetonitrile and 2,6-lutidine mixture, 20% N-methylimidazole (NMI) in acetonitrile as capping agent A, isobutryic anhydride in a 1 :1 mixture of acetonitrile and 2,6-lutidine mixture as capping agent B and 20% DEA in acetonitrile.
The oligonucleotide synthesis was performed in the direction of 5′ to 3′ utilizing a repetitive synthesis cycle consisting of detritylation followed by coupling, and sulfurization performed at ambient temperature.
A column (diameter : 3.5 cm) was packed with a solid-support loaded with 3-palmitoylamido-1-0-(4, 4′-dimethoxytrityl)-2-0-succinyl propanediol (3.5 mmol based on a capacity of 400 μιηοΙ/g) that was coupled with the nucleotide monomer B Detritylation was achieved using 3% dichloroacetic acid (DCA) in toluene (amount is between 6.5 and 13.4 column volumes in each detritylation step) and the solid-support bound nucleotide was washed with acetonitrile (amount : 5 column volumes). Coupling with the next nucleotide monomer of formula (B n) was achieved by pumping a solution of 0.5 M 5-(ethylthio)-1 H-tetrazole in acetonitrile and 0.15 M of the next nucleotide monomer of formula (B n) in the sequence, dissolved in acetonitrile, through the column. The column was washed with acetonitrile (amount : 2 column volumes). Then sulfurization was performed by pumping a solution of 0.2 M phenyl acetyl disulfide (PADS) in a 1 :1 mixture of acetonitrile and 2,6-lutidine mixture through the column followed by washing the column with acetonitrile (amount :
5 column volumes).
The sulfurization was followed by a capping step. Each capping in a given cycle used 37-47 equivalents (eq.) of the capping agent NMI, and 9-1 1 equivalents of the capping agent B isobutryic anhydride (IBA), and 1 .4-1.8 equivalents of 2,6 lutidine. Capping agents A and B were pumped through the column with separate pumps at different ratios such as 50:50, 35:65, 65:35.
The synthesis cycle of detritylation, coupling with the next nucleotide monomer of formula (B n) and sulfurization, and capping step was repeated 12 times, followed by detritylation using 3% dichloroacetic acid (DCA) in toluene (amount is between 6.5 and 13.4 column volumes).
Upon completion of the synthesis cycle, the crude oligonucleotide on the solid-support support was treated with a diethylamine (DEA) solution followed by treatment with ammonium hydroxide solution: ethanol (3: 1 volume ratio) at a temperature of 55°C. The reaction mixture was aged for 4 to 24 hours at 55°C, cooled to room temperature, and slurry was filtered to remove the polymeric support. The solution comprising imetelstat in its ammonium form was subjected to the HPLC analysis procedure of Experiment 3.
Experiment 3 : comparision of no-capping vs. capping
Imetelstat obtained in Experiment 1 and Experiment 2 was analysed by HPLC. The amount of the desired full length oligonucleotide having 13 nucleotides was determined and listed in the Table below for Experiment 1 and Experiment 2. Also, the total amount of shortmer, specifically the 12mer, was determined and listed in the Table below for Experiment 1 and Experiment 2.
HPLC analysis method :
column type: Kromasil C18, 3.5 μιτι particle size, 4.6 X 150 mm
eluent:
A: 14.4 mM TEA/386 mM HFIP (hexafluoroisopropanol) /100 ppm(w/v) Na2EDTA in water B: 50% MeOH, 50% EtOH containing 5% IPA
Gradient :
Step Run time (minutes) %B
1 0 10
2 5 10
3 12 26 (linear)
4 35 45 (linear)
5 40 50 (linear)
6 42 50
7 44 10 (linear)
8 50 10
Table : capping vs. no-capping experiments (Experiment 1 was run twice and results are listed as Experiment 1a and 1 b).
The HPLC analysis of Experiment 1 and Experiment 2 demonstrates that yield and purity are comparable for the no-capping experiment vs. the capping experiment.
Main peak % includes Full length oligonucleotide + PO impurities + depurinated impurities.
PO impurities are impurities including one or more oxophosphoramidate internucleoside linkages instead of thiophosphoramidate internucleoside linkages.
Solvent use and reaction time
0.45 L of acetonitrile/mmol is used to prepare capping agent A and capping agent B reagents which corresponds to approximately 25 % of the overall acetonitrile use during the preparation of the reagents. Since each chemical reaction step is followed by a solvent wash, after each capping step too, a solvent wash takes place which is equivalent to about 40 column volumes of the solvent. Considering that about 212 column volumes of the solvent wash is done for a given synthesis run, about 19 % of the wash solvent is used for the capping steps. Each capping step takes between 3 – 6 minutes. This corresponds to about 8 % of the overall synthesis time including the 13 cycles and DEA treatment.
Experiment 4 (detritylation temperature)
The detritylation temperature has an impact in terms of controlling n-1 and depurinated impurities. The temperature of the deblocking solution at the entrance of the synthesizer was chosen between 17.5 and 27 °C (at 3.5 mmol scale) and the selected temperature was kept the same for all detritylation steps. The acetonitrile washing was also kept at the same temperature of the deblocking solution. The % depurinated impurities increased linearly with temperature while n-1 was higher at lower temperatures.
Temperature n-1 % Depurinated Impurity %
17.5 10.7 5.3
19 7.6 6.4
22 5.4 8.7
25 6.1 10.8
27 5.3 12.3
Experiment 5 (sulfurization step temperature)
In the experiments below, the temperature (RT means room temperature) of the PADS solution used in the sulfurization reactions was tested for the % of less favourable PO impurities (these are impurities where phosphor oxidation occurred instead of sulfurization). Lower temperature results in lower PO %.
SEQ ID NO:1 – imetelstat and imetelstat sodium
5′-R-TAGGGTTAGACAA-NH2-3′
wherein R represents palmitoyl [(CH2)1 CH3] amide is conjugated through an aminoglycerol linker to the 5′-thiophosphate group of an N3′ – P5′ thiophosphoramidate (NPS) -linked oligonucleotide.
///////////IMETELSTAT, GRN163L, PHASE 3, orphan drug, FAST TRACK
CCCCCCCCCCCCCCCC(=O)NCC(COP(=S)([O-])OCC1C(CC(O1)N2C=C(C(=O)NC2=O)C)NP(=S)([O-])OCC3C(CC(O3)N4C=NC5=C4N=CN=C5N)NP(=S)([O-])OCC6C(CC(O6)N7C=NC8=C7N=C(NC8=O)N)NP(=S)([O-])OCC9C(CC(O9)N1C=NC2=C1N=C(NC2=O)N)NP(=S)([O-])OCC1C(CC(O1)N1C=NC2=C1N=C(NC2=O)N)NP(=S)([O-])OCC1C(CC(O1)N1C=C(C(=O)NC1=O)C)NP(=S)([O-])OCC1C(CC(O1)N1C=C(C(=O)NC1=O)C)NP(=S)([O-])OCC1C(CC(O1)N1C=NC2=C1N=CN=C2N)NP(=S)([O-])OCC1C(CC(O1)N1C=NC2=C1N=C(NC2=O)N)NP(=S)([O-])OCC1C(CC(O1)N1C=NC2=C1N=CN=C2N)NP(=S)([O-])OCC1C(CC(O1)N1C=CC(=NC1=O)N)NP(=S)([O-])OCC1C(CC(O1)N1C=NC2=C1N=CN=C2N)NP(=O)(OCC1C(CC(O1)N1C=NC2=C1N=CN=C2N)N)[S-])O.[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+]