














LANRAPLENIB
GS-9876
Phase II, GILEAD
| Phase II | Gilead | Cutaneous lupus erythematosus
Rheumatoid arthritis Sjogren syndrome |
| GS-9876 | ||
| LANRAPLENIB |
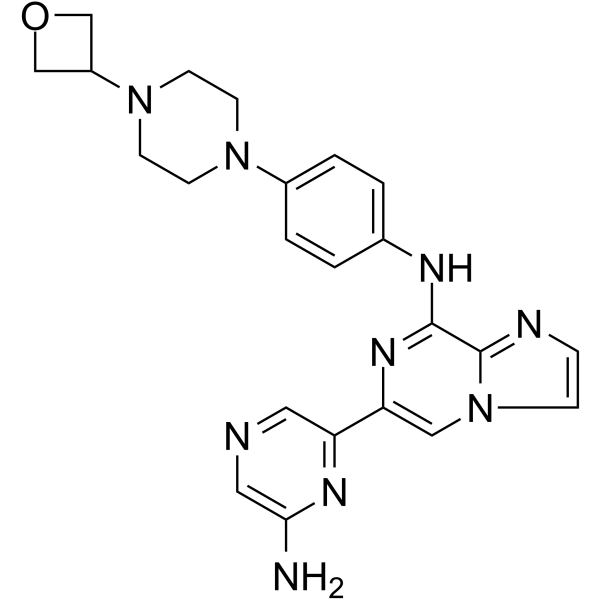
Imidazo(1,2-a)pyrazin-8-amine, 6-(6-amino-2-pyrazinyl)-N-(4-(4-(3-oxetanyl)-1-piperazinyl)phenyl)-
6-(6-Aminopyrazin-2-yl)-N-(4-(4-(oxetan-3-yl)piperazin-1-yl)phenyl)imidazo|1,2-a]pyrazin-8-amine
6-(6-Amino-2-pyrazinyl)-N-(4-(4-(3-oxetanyl)-1-piperazinyl)phenyl)imidazo(1,2-a)pyrazin-8-amine
| Molecular Weight |
443.50 |
|---|---|
| Formula |
C₂₃H₂₅N₉O |
| CAS No. |
1800046-95-0 |
Lanraplenib (GS-9876) is a highly selective and orally active SYK inhibitor (IC50=9.5 nM) in development for the treatment of inflammatory diseases. Lanraplenib (GS-9876) inhibits SYK activity in platelets via the glycoprotein VI (GPVI) receptor without prolonging bleeding time (BT) in monkeys or humans.
| Description |
Lanraplenib (GS-9876) is a highly selective and orally active SYK inhibitor (IC50=9.5 nM) in development for the treatment of inflammatory diseases. Lanraplenib (GS-9876) inhibits SYK activity in platelets via the glycoprotein VI (GPVI) receptor without prolonging bleeding time (BT) in monkeys or humans[1][2][3]. |
|---|---|
| IC50 & Target |
IC50: 9.5 nM (SYK)[1] |
| In Vitro |
Lanraplenib (GS-9876) inhibits anti-IgM stimulated phosphorylation of AKT, BLNK, BTK, ERK, MEK, and PKCδ in human B cells with EC50 values of 24-51 nM. Lanraplenib (GS-9876) inhibits anti-IgM mediated CD69 and CD86 expression on B-cells (EC50=112±10 nM and 164±15 nM, respectively) and anti-IgM /anti-CD40 co-stimulated B cell proliferation (EC50=108±55 nM). In human macrophages, Lanraplenib (GS-9876) inhibits IC-stimulated TNFα and IL-1β release (EC50=121±77 nM and 9±17 nM, respectively)[1]. |

Lanraplenib succinate
1800047-00-0
UNII-QJ2PS903VZ
QJ2PS903VZ
GS-SYK Succinate
1241.3 g/mol, C58H68N18O14
6-(6-aminopyrazin-2-yl)-N-[4-[4-(oxetan-3-yl)piperazin-1-yl]phenyl]imidazo[1,2-a]pyrazin-8-amine;butanedioic acid
PAPER
https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00621
https://pubs.acs.org/doi/suppl/10.1021/acsmedchemlett.9b00621/suppl_file/ml9b00621_si_001.pdf

Spleen tyrosine kinase (SYK) is a critical regulator of signaling in a variety of immune cell types such as B-cells, monocytes, and macrophages. Accordingly, there have been numerous efforts to identify compounds that selectively inhibit SYK as a means to treat autoimmune and inflammatory diseases. We previously disclosed GS-9973 (entospletinib) as a selective SYK inhibitor that is under clinical evaluation in hematological malignancies. However, a BID dosing regimen and drug interaction with proton pump inhibitors (PPI) prevented development of entospletinib in inflammatory diseases. Herein, we report the discovery of a second-generation SYK inhibitor, GS–9876 (lanraplenib), which has human pharmacokinetic properties suitable for once-daily administration and is devoid of any interactions with PPI. Lanraplenib is currently under clinical evaluation in multiple autoimmune indications.
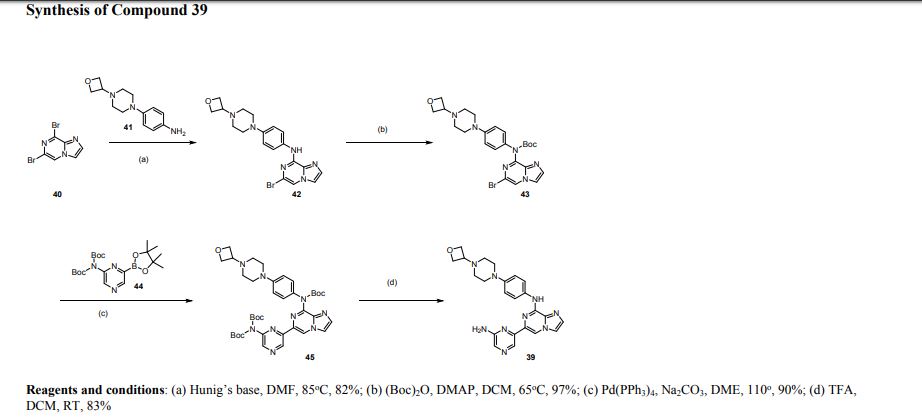
Step 6. 6-(6-Aminopyrazin-2-yl)-N-(4-(4-(oxetan-3-yl)piperazin-1-yl)phenyl)imidazo|1,2-a]pyrazin-8-amine (39). To a solution of tert-butyl(6-(6-(bis(tert-butoxycarbonyl)amino)pyrazm-2-yl)imidazo[1,2-a]pyrazin-8-yl)(4-(4-(oxetan-3-yl)piperazin1yl)phenyl)carbamate 45 (200 mg, 0.269 mmol) in DCM (2 ml) was added TFA (0.5 ml, 6.578 mmol). The reaction was stirred at room temperature for 16h, treated with saturated sodium bicarbonate, extracted with EtOAc, and purified on silica gel, eluting with 5%MeOH / EtOAc to 20%MeOH / EtOAc. The desired fractions were combined and concentrated to provide 100 mg (83% yield) of the title compound 39. m/z calcd for C23H25N9O [M+H] + 444.23, found LCMS-ESI+ (m/z): [M+H] + 444.20. 1H NMR (300 MHz d6-DMSO) δ: 9.5 (s,lH), 8.588 (s, 1H), 8.47 (s, 1H), 8.12 (d, 1H), 7.95-7.92 (d5 2H), 7.88 (s, 1H), 7.62 (s, 1H), 6.99-6.96 (d, 2H), 6.46 (s, 2H), 4.57- 4.53 (m, 2H), 4.48-4.44 (m, 2H), 3.43 (m, 1H), 3.15-3.12 (m, 4H), 2.41- 2.38 (m, 4H).
MORE SYNTHESIS COMING, WATCH THIS SPACE…………………..

SYNTHESIS
PATENT
WO 2015100217
WO 2016010809
PATENT
WO 2016172117
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016172117
Protein kinases, the largest family of human enzymes, encompass well over 500 human proteins. Spleen Tyrosine Kinase (Syk) is a member of the Syk family of tyrosine kinases, and is a regulator of early B-cell development as well as mature B-cell activation, signaling, and survival.
Acute Graft Versus Host Disease (aGVHD), also known as fulminant Graft Versus Host Disease, generally presents symptoms within the first 100 days following allogenic hematopoietic stem cell transplantation and is generally characterized by selective damage to the skin, liver, mucosa, and gastrointestinal tract. Chronic Graft Versus Host Disease (cGVHD) occurs in recipients of allogeneic hematopoietic stem cell transplant (HSCT). GVHD is considered chronic when it occurs >100 days post-transplant, though aspects of cGVHD may manifest themselves prior to the 100 day point and overlap with elements of aGVHD. The disease has a cumulative incidence of 35-70% of transplanted patients, and has an annual incidence of approximately 3,000-5,000 and a prevalence of approximately 10,000 in the US. cGVHD is difficult to treat and is associated with worse outcomes compared to those without cGVHD. Current standard of care includes a variety of approaches including systemic corticosteroids often combined with calcineurin inhibitors, mTOR inhibitors, mycophenylate mofetil, or rituximab. Despite treatment, response rates are poor (40-50%) and cGVHD is associated with significant morbidity such as serious infection and impaired quality of life; the 5-year mortality is 30-50% (Blazar et al., Nature Reviews Immunology 12, 443-458, June 2012).
Human and animal models have demonstrated that aberrant B-lymphocyte signaling and survival is important in the pathogenesis of cGVHD. B-cell targeted drugs, including SYK inhibitors (fostamatinib – Sarantopoulos et al, Biology of Blood and Marrow Transplantation, 21(2015) S 11 -S 18) and BTK inhibitors (ibrutinib – Nakasone et al, Int. J. HematoL- 27 March 2015), have been shown to selectively reduce the function and frequency of aberrant GVHD B-cell populations ex vivo.
There remains a need for new methods, pharmaceutical compositions, and regimens for the treatment of GVHD, including aGVHD and cGVHD.
Example 2. Preparation of 6-(6-aminopyrazin-2-yl)-N-(4-(4-(oxetan-3-yl)piperazin-l- yl)phenyl)imid azo [ 1,2-a] pyrazin-8-amine (2)
2-Bis(tert-butoxycarbonyl)amino-6-bromopyrazine XIV: To a mixture of 6-bromopyrazin-2-amine (5 g, 28.7 mmol) and di-tert-butyl dicarbonate (25.09 g, 1 14.94 mmol) was added DCM (10 ml) followed by DMAP (0.351 g, 29 mmol). The reaction was heated to 55 °C for lh, cooled to RT, the reaction was partitioned between water and DCM, purified on silica gel and concentrated to provide of 2-bis(tert-butoxycarbonyl)amino-6-bromopyrazine XIV. LCMS-ESI+ (m/z): [M+H]+: 374.14. XH NMR (DMSO) δ: 8.84(d, 2H), 1.39 (s, 18H).
tert-Butyl (6-(6-(bis(tert-butoxycarbonyl)amino)pyrazin-2-yl)imidazo[l,2-a]pyrazin-8-yl)(4-(4-(oxetan-3-yl)piperazin-l-yl)phenyl)carbamate XVI – CHEMISTRY A route: tert-Butyl 4-(4-(oxetan-3-yl)piperazin-l-yl)phenyl(6-(tributylstannyl)imidazo[l,2-a]pyrazin-8-yl)carbamate V (215 mg, 0.291 mmol), was combined with 2-bis(tert-butoxycarbonyl)amino-6-bromopyrazine XIV (217.58 mg, 0.581 mmol),
bis(triphenylphosphine)palladium(II) dichloride(30.61 mg, 0.044 mmol) and 1,4-dioxane (5ml). The reaction mixture was stirred in a microwave reactor at 120 °C for 30 min. The reaction mixture was quenched with saturated KF, extracted with EtOAc, purified on silica gel, eluted with EtOAc. The desired fractions were combined and concentrated to provide 100 mg (46% yield) of tert-butyl (6-(6-(bis(tert-butoxycarbonyl)amino)pyrazin-2-yl)imidazo[l,2-a]pyrazin-8-yl)(4-(4-(oxetan-3-yl)piperazin-l-yl)phenyl)carbamate XVI. LCMS-ESI+ (m/z): [M+H]+: 744.4. lH NMR (300 MHz d6-DMSO) δ: 9.37 (s, 1H), 9.18 (s, 1H), 8.77 (s, 1H), 8.33 (d, 1H), 7.87 (d, 1H), 7.28-7.25 (d, 2H), 6.92-6.89 (d, 2H), 4.55-4.41 (m, 4H), 3.4 (m, lH), 3.14-3. 11 (m,4H), 2,37-2.34 (m, 4H), 1.37 (s, 18H), 1.3 (s, 9H).
tert-Butyl (6-(6-(bis(tert-butoxycarbonyl)amino)pyrazin-2-yl)imidazo[l,2-a]pyrazin-8-yl)(4-(4-(oxetan-3-yl)piperazin-l-yl)phenyl)carbamate XVI – CHEMISTRY B route: Step 1 : To a dry 250 mL round-bottomed flask was added 2-bis(tert-butoxycarbonyl)amino-6-bromopyrazine XIV (l .Og, l .Oequiv, 2.67mmol), KOAc (790mg, 8.02mmol, 3.0equiv), 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(l ,3,2-dioxaborolane) (750mg, 2.94mmol, l . l equiv), Pd(dba) (171mg, 0.187mmol, 0.07equiv) and X-phos (128mg, 0.267mmol, O. lequiv) followed by 1,4-dioxane (25mL) and the solution was sonicated for 5 min and then purged with N2 gas for 5 min. The flask with contents was then placed under N2 atmosphere and heated at 1 10 °C for 90 min. Once full conversion to the pinacolboronate was achieved by LCMS, the reaction was removed from heat and allowed to cool to RT. Once cool, the reaction contents were filtered through Celite and the filter cake was washed 3 x 20 mL EtOAc. The resultant solution was then concentrated down to a deep red-orange
syrup providing N, N-BisBoc 6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyrazin-2-amine XV, which was used directly in the next step.
Step 2: The freshly formed N, N-BisBoc 6-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)pyrazin-2-amine XV (2.67 mmol based on 100% conversion, 2.0 equiv based on bromide) was dissolved in 20 Ml of 1,2-dimethoxy ethane and to that solution was added tert-butyl (6-bromoimidazo[l,2-a]pyrazin-8-yl)(4-(4-(oxetan-3-yl)piperazin-l-yl)phenyl)carbamate IV (707mg, 1.34mmol, l .Oequiv), Na2CC>3 (283mg, 2.67mmol, 2.0equiv), Pd(PPh3)4 (155mg, 0.134mmol, 0.1 equiv) and water (l OmL) and the solution was degassed for 5 min using N2 gas. The reaction was then placed under N2 atmosphere and heated at 110 °C for 90 min. LCMS showed complete consumption of the bromide starting material and the reaction was removed from heat and allowed to cool to RT. The reaction was diluted with 100 mL water and 100 mL 20% MeOH/DCM and the organic layer was recovered, extracted 1 x sat. NaHCCb, 1 x sat brine and then dried over Na2SC>4. The solution was then filtered and concentrated down to an orange-red solid. The sample was then slurried in warm MeOH, sonicated then filtered, washing 2 x 20 mL with cold MeOH and then the cream-colored solid was dried on hi-vacuum overnight to yield 905 mg of tert-butyl (6-(6-(bis(tert-butoxycarbonyl)amino)pyrazin-2-yl)imidazo[l,2-a]pyrazin-8-yl)(4-(4-(oxetan-3-yl)piperazin- 1 -yl)phenyl)carbamate XVI.
6-(6-Aminopyrazin-2-yl)-N-(4-(4-(oxetan-3-yl)piperazin-l-yl)phenyl)imidazo[l,2-a]pyrazin-8-amine (2): To a solution of tert-butyl (6-(6-(bis(tert-butoxycarbonyl)amino)pyrazin-2-yl)imidazo[l,2-a]pyrazin-8-yl)(4-(4-(oxetan-3-yl)piperazin-l -yl)phenyl)carbamate XVI (200 mg, 0.269 mmol) in DCM (2 ml) was added TFA (0.5 ml, 6.578 mmol). The reaction was stirred at rt for 16h, saturated sodium bicarbonate was added, extracted with EtOAC and purified on silica gel, eluted with 5%MeOH / EtOAc, 20%MeOH / EtOAc. The desired fractions were combined and concentrated to provide the title compound 2. LCMS-ESI+(m/z): [M+H]+: 444.2. lH NMR (300 MHz d6-DMSO) δ: 9.5 (s, lH), 8.588 (s, IH), 8.47 (s, IH), 8. 12 (d, IH), 7.95-7.92 (d, 2H), 7.88 (s, IH), 7.62 (s, IH), 6.99-6.96 (d, 2H), 6.46 (s, 2H), 4.57-4.53 (m, 2H), 4.48-4.44 (m, 2H), 3.43 (m, IH), 3.15-3.12 (m, 4H), 2.41 -2.38 (m, 4H).
Example 2 – Alternate Synthesis
H2S04, water
Di-feri-butyl {6-[8-({4-[4-(oxetan-3-yl)piperazin-l-yl]phenyl}amino)imidazo[l,2-fl]pyrazin-6-yl]pyrazin-2-yl}imidodicarbonate:
To a 720 L reactor, was added di-fer/-butyl (6-bromopyrazin-2-yl)imidodicarbonate (18.5 kg, 1.41 equiv, 49 mol), bis(pinacolato)diboron (13.8 kg, 1.56 equiv, 54 mol), potassium propionate (11.9 kg, 3.02 equiv, 106 mol), and bis(di-fer/-butyl(4-dimethylaminophenyl) phosphine)dichloropalladium (1.07 kg, 0.0043 equiv, 1.5 mol), followed by degassed toluene (173 L). The mixture was degassed then heated at 65 °C until the reaction was deemed complete (0% tert-butyl 2-((6-bromopyrazin-2-yl)(tert-butoxycarbonyl)amino)-2-oxoacetate) by UPLC. Upon completion, the reaction was cooled to 23 °C. Once cooled, 6-bromo-N-(4-(4-(oxetan-3-yl)piperazin-l -yl)phenyl)imidazo[l ,2-a]pyrazin-8-amine (15.0 kg, 1.00 equiv, 35 mol) was added and the mixture was degassed. A degassed aqueous potassium carbonate solution prepared using water (54 L) and potassium carbonate (20.6 g, 4.26 equiv, 149 mol) was then added to the reaction mixture and the reactor contents was degassed. The reactor contents was heated at 65 °C until reaction was deemed complete (1% 6-bromo-N-(4-(4-(oxetan-3-yl)piperazin-l-yl)phenyl)imidazo[l,2-a]pyrazin-8-amine) by UPLC. Upon completion, the reaction was cooled to 24 °C.
The cooled mixture was concentrated and then diluted with dichloromethane (300 L), transferred to a 1900 L reactor and rinsed forward with dichloromethane (57 L). N-acetyl-L-cysteine (3.8 kg) was charged and the mixture was agitated for 15 h. Water (135 L) was then added and the mixture was filtered and rinsed forward with dichloromethane (68 L). The organic layer was recovered and washed with a brine solution prepared using water (68 L) and sodium chloride (7.5 kg).
The resultant organic layer was polish filtered then concentrated and fert-butyl methyl ether (89.9 kg) was slowly charged keeping the temperature at 31 °C. The contents was cooled to 0 °C and aged, then filtered and rinsed with tert-butyl methyl ether (32.7 kg) and dried at 40 °C to give 17.2 kg of di-tert-butyl {6-[8-({4-[4-(oxetan-3-yl)piperazin-l-yl]phenyl} amino)imidazo[l,2-a]pyrazin-6-yl]pyrazin-2-yl}imidodicarbonate.
LCMS-ESf (m/z): [M+H]+: 644.3. ΧΗ ΝΜΚ (400 MHz, CDC13) δ: 9.43 (s, 1H), 8.58 (s, 1H), 8.53 (s, 1H), 8.02 (s, 1H), 7.84 (m, 2H), 7.63 (d, 1H), 7.61 (d, 1H), 7.04 (m, 2H), 4.71 (m,4H), 3.59 (m, lH), 3.27 (m, 4H), 2.55 (m, 4H), 1.46 (s, 18H).
6-(6-Aminopyrazin-2-yl)-N-(4-(4-(oxetan-3-yl)piperazin-l-yl)phenyl)imidazo[l,2-a]pyrazin-8-amine succinate (Example 2):
To a slurry of di-tert-butyl {6-[8-({4-[4-(oxetan-3-yl)piperazin-l -yl]phenyl} amino)imidazo[l,2-a]pyrazin-6-yl]pyrazin-2-yl}imidodicarbonate (225 g, 0.35 mol, 1 mol eq.) in water (12 parts) was added a solution of sulfuric acid (3.1 parts, 6.99 mol, 20 mol eq.) in water (5 parts). The reaction was heated to ca. 40 °C and stirred at this temperature for ca. 4 h at which point the reaction is deemed complete. The reaction mixture was cooled to ca. 22 °C, acetone (3 parts) was charged and a solution of sodium carbonate (4.1 parts, 8.75 mol, 25.0 mol eq.) in water (15 parts) was added. The resulting slurry was filtered and the wet cake was washed with water in portions (4 x 1 parts), then with fert-butyl methyl ether (4 parts). The wet cake (Example 2 free base) was dried at ca. 60 °C. To the slurry of dry Example 2 free base in 2-propanol (2.3 parts) was added a solution of succinic acid (Based on the isolated Example 2 free base: 0.43 parts, 1.6 mol eq.) in 2-propanol (15 parts). The resulting slurry was heated to ca. 40 °C and stirred at this temperature for ca. 2 h and then cooled to ca. 22 °C, followed by a stir period of ca. 16 h. The slurry was filtered at ca. 22 °C and the wet cake was washed with 2-propanol (5 parts) and dried at ca. 60 °C to afford the product.
LCMS-ESI+ (m/z): [M+H]+: 620.65. ¾ NMR (400 MHz d6-DMSO) δ: 12.2 (broad s, 1.5H), 9.58 (s, IH), 8.63 (s, IH), 8.50 (s, IH), 8.15 (s, IH), 7.95 (d, 2H), 7.90 (s, IH), 7.64 (s, IH), 7.00 (d, 2H), 6.50 (s, 2H), 4.52 (dd, 4H), 3.45 (m, IH), 3.19 (m, 4H), 2.40 (m, 10H).
REF
/////////////LANRAPLENIB, GS-9876, SYK inhibitor
NC1=CN=CC(C2=CN3C(C(NC4=CC=C(N5CCN(C6COC6)CC5)C=C4)=N2)=NC=C3)=N1