














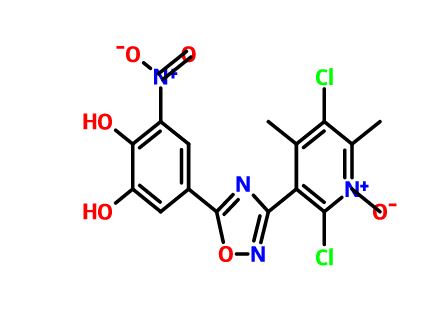
Opicapone
2,5-dichloro-3-(5-(3,4-dihydroxy-5-nitrophenyl)-1,2,4-oxadiazol-3-yl)-4,6-dimethylpyridine-1-oxide
BIA-9-1067; ONO-2370; BIA-91067
CAS No.923287-50-7
MF C15H10Cl2N4O6
MW: 411.9977
TRADE NAME (Ongentys®)
Approved EU 2016-06-24 BIAL PORTELA
| PORTELA & CA. S.A. [PT/PT]; Av. Da Siderurgia Nacional, P-4745-457 S. Mamede do Coronado (PT) |
LEARMONTH, David Alexander; (PT).
KISS, Laszlo Erno; (PT).
LEAL PALMA, Pedro Nuno; (PT).
DOS SANTOS FERREIRA, Humberto; (PT).
ARAÚJO SOARES DA SILVA, Patrício Manuel Vieira; (PT)
MOA:Catechol-O-methyl transferase (COMT) inhibitor
Indication:Parkinson’s disease (PD)
A COMT inhibitor used as adjunctive therapy for parkinson’s disease.

Opicapone, also known as BIA 9-1067, is a novel potent and selective catechol-O-methyltransferase inhibitor (COMT inhibitor ) under clinical evaluation as an adjunct to L-Dopa therapy of Parkinson’s disease. Opicapone, a novel third generation COMT inhibitor, when compared to entacapone, provides a superior response upon the bioavailability of levodopa associated to more pronounced, long-lasting, and sustained COMT inhibition
Opicapone was approved by European Medicine Agency (EMA) on Jun 24, 2016. It was developed and marketed as Ongentys® by Bial – Portela in EU.
Opicapone is a selective and reversible COMT inhibitor, used as adjunctive therapy for Parkinson’s disease.
Ongentys® is available as hard capsules, containing 25 mg and 50 mg of opicapone. The recommended dose is 50 mg, taken once a day at bedtime, at least one hour before or after levodopa combination medicines.
Catechol-O-methyltransferase (COMTa) catalyzes the transfer of a methyl group from S-adenosyl-l-methionine to catecholic substrates such as endogenous catechol neurotransmitters(2)and xenobiotics including (S)-2-amino-3-(3,4-dihydroxyphenyl)propanoic acid (l-Dopa), the gold standard drug for treatment of Parkinson’s disease (PD). Coadministration of a peripheral amino acid decarboxylase (AADC) inhibitor prevents breakdown of l-Dopa in the periphery by blocking enzymatic decarboxylation, and inhibition of COMT further improves its bioavailability by reducing the formation of 3-O-methyl-l-Dopa (3-OMD).
Abbreviations: COMT, catechol-O-methyltransferase; PD, Parkinson’s disease; AADC, amino acid decarboxylase; SAR, structure−activity relationship; ADMET, absorption, distribution, metabolism, excretion, toxicity; l-Dopa, (S)-2-amino-3-(3,4-dihydroxyphenyl)propanoic acid; 3-OMD, 3-O-methyl-l-Dopa.
![]()

Chemical structures of tolcapone 1, entacapone 2, and nebicapone 3.
Opicapone
A preferred method of treatment of Parkinson’s disease is the administration of a combination of levodopa and a peripherally selective aromatic amino acid decarboxylase inhibitor (AADCI) together with a catechol-O-methyltransferase (COMT) inhibitor. The currently employed COMT inhibitors are tolcapone and entacapone. However, some authorities believe that each of these COMT inhibitors have residual problems relating to pharmacokinetic or pharmacodynamic properties, or to clinical efficiency or safety. Hence, not all patients get most benefit from their levodopa/AADCI/COMT inhibitor therapy.
Favoured new COMT inhibitors were disclosed in L. E. Kiss et al, J. Med. Chem., 2010, 53, 3396-3411 (D1), WO 2007/013830 (D2) and WO 2007/117165 (D3) which are believed to have particularly desirable properties so that patients can benefit from enhanced therapy.
D1, D2 and D3 also disclosed methods of preparing the new COMT inhibitors. Those processes, although effective, would benefit from an increase in yields. Other benefits which would be appropriate include those selected from reduction in number of process steps, reduction in number of unit operations, reduction of cycle-times, increased space yield, increased safety, easier to handle reagents/reactants and/or increase in purity of the COMT inhibitor, especially when manufacture of larger quantities are envisaged. A process has now been discovered that proceeds via a new intermediate which is suitable for manufacture of commercially useful quantities of a particularly apt COMT inhibitor in good yield. Additional benefits occur such as those selected from a reduced number of process steps and number of unit operations, reduced cycle-times, increased space yield, increased safety, with easier to handle reagents/reactants, improved impurity profile and/or good purity.
CLINICAL
https://clinicaltrials.gov/show/NCT01851850

SYN1
Discovery of a Long-Acting, Peripherally Selective Inhibitor of Catechol-O-methyltransferase

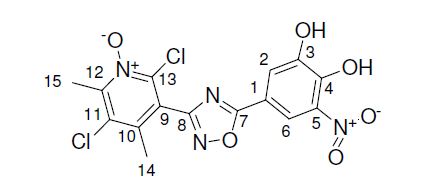
1H : 11.07 (2H, br, OH), 8.11 (1H, d, J = 2 Hz, H6), 7.73 (1H, d, J = 2 Hz, H2), 2.66 (3H, s, H15),2.24 (3H, s, H14).
(C15H10Cl2N4O6) C, H, N, S: Calc: C, 43.60; H, 2.44; N, 13.56; Found: C, 44; H, 2.3; N, 13.6.

PATENT
LEARMONTH, David Alexander; (PT).
KISS, Laszlo Erno; (PT).
LEAL PALMA, Pedro Nuno; (PT).
DOS SANTOS FERREIRA, Humberto; (PT).
ARAÚJO SOARES DA SILVA, Patrício Manuel Vieira; (PT)
PATENT
The present invention in one aspect provides 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4,oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene and salts thereof, that is the compound of the formula (I):

and salts thereof.
Most aptly the compound of formula (I) is unsalted. However, salts of the hydroxy group with metal ions such as the alkali or alkaline earth metals, particularly the sodium and potassium salts are provided as well as those of highly basic organic compounds such as guanidine or the like.
Particularly suitably the compound of formula (I) or its salt is provided in a form suitable for use as a chemical intermediate. This may be, for example, in a form at least 50% pure, in crystalline form, in solid form or in an organic solvent or the like.
The compound of formula (I) is useful as an intermediate in the preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol i.e. the compound of formula II):

The compound of formula (II) may also be referred to as opicapone or 2,5-dichloro-3-(5-(3,4-dihydroxy-5-nitrophenyl)-[1,2,4]-oxadiazole-3-yl)-4,6-dimethylpyridine-1-oxide. Opicapone has been found to be more potent than tolcapone in inhibiting liver COMT both at 3 hours and 6 hours post oral administration to rats [ED50 in mg/kg, opicapone 0.87 at 3 hours and 1.12 at 6 hours as compared to tolcapone 1.28 at 3 hours and 2.08 at 6 hours]. Opicapone at a dose of 3 mg/kg was found to be more effective at inhibiting rat liver COMT with nearly complete inhibition occurring 2 to 6 hours post oral administration with only about 90% of enzyme activity recovered after 72 hours while tolcapone provided shorter duration of activity with about 84% recovery after only 9 hours. Both opicapone and tolcapone inhibit human recombinant S-COMT but opicapone has an inhibitory constant of 16pM being 10 fold lower than that for tolcapone. With respect to the desirable property of avoiding inhibition of COMT in the brain, opicapone following oral administration to the rat was found to be devoid of effect whereas tolcapone inhibited about 50% of enzyme activity over a period of 8 hours post administration.
Preparation 1

Cyanoacetamide (280g) was reacted with acetyl acetone (352.9g) in methanol (1015g) and morpholine (14.9g). The reaction was stirred under reflux at 65 °C until the reaction appeared complete. The resulting product suspension was filtered, washed with methanol and dried to provide the desired product about 97% yield.
Preparation 2

The product of Preparation 1 (159g) was suspended in acetonitrile (749.5g) and cooled to 0-5°C. Sulfuryl chloride (178.9g) was added and the reaction mixture warmed to room temperature and stirred until the reaction appeared complete.
The resulting suspension is cooled to 0-5°C and filtered. The solid was washed with acetonitrile, ethyl acetate and heptane. The product was then dried under vacuum at 50°C to yield the desired product (82%).
Preparation 3

Phosphoryl chloride (973.2g), tetramethylammonium chloride (67.3g) and compound of Preparation 2 (227.1g) were added to dichloromethane (500g). The suspension was heated to 85°C and stirred for 5 hours. Excess of phosphoryl chloride was removed by distillation in vacuo. The reaction mixture was cooled below 30°C and diluted with dichloromethane. The resulting solution was added to water (1350g) at room temperature and stirred for 30 minutes. The lower organic phase was separate and the aqueous phase extracted with dichloromethane. The organic phases were combined, washed with water and then treated with charcoal. The charcoal was filtered and a solvent swap to heptane was performed by distillation at atmospheric pressure. The solution was filtered at 50°C and then cooled to 30°C. On further cooling to 0°C
crystals were obtained. These were isolated by filtration, washed twice with heptane. After drying at 50°C the desired product was obtained typically at 88-91 % .
The above process was repeated with a reduction in dichloromethane during crystallisation and adding some methanol. The resulting plate-like crystals were more easily transferred for subsequent use.
Preparation 4a

Product of Preparation 3 (68.6g) and 1,10-phenanthroline monohydrate (0.9g) were suspended in methanol (240g) at room temperature. Water (518g) and a hydroxylamine solution (50% in water, 80.9g), were added and the mixture heated to 70-80°C and stirred for 5-6 hours. Water was added at 70-80 °C and the solution held for 1 hour to induce crystallization. Crystallization was completed by cooling to 15°C over 8 hours. The product was filtered off and washed twice with water and dried at 50°C under vacuum. The product was an off white to light yellow and the yield was 87.9% .
Preparation 4b

A suspension of 2,5-Dichloro-4,6-dimethyl-nicotinonitrile (45.0 kg) and 50% hydroxylamine (59.2 kg) in the presence of catalytic amount of 1,10-phenanthroline monohydrate (0.680 kg) in methanol / water (214 kg/362 kg) is heated to 70-80°C. The mixture is agitated at 70-80°C. Water (353 kg) is added slowly into the resulting solution while the temperature is maintained at > 79°C. The solution is cooled to 75 °C with stirring resulting in crystallization of (Z)-2,5-dichloro-N’-hydroxy-4,6-
dimethylnicotinimidamide. The suspension is further cooled to 20 °C, the solid is filtered off and the wet cake is washed with water (160 kg). (Z)-2,5-dichloro-N’-hydroxy-4,6-dimethylnicotinimidamide is dried under vacuum at max. 60°C until residual water level is max 0.15% (KF).
Example 1a
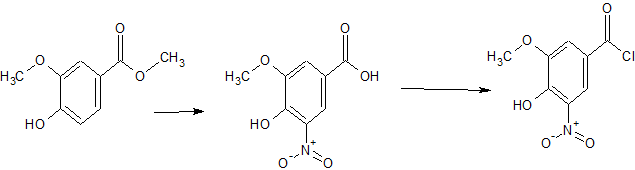
Preparation of 4-hydroxy-5-methoxy-3 nitrobenzoic acid

Vanillic acid (75g) was suspended in acetic acid (788g). The suspension was cooled to 10°C to 15°C and nitric acid (49g or 65% solution) was added over three hours at a rate which kept temperature between 10°C and 20°C. The resulting yellow orange was stirred for a further one hour at 18°C to 23°C. The suspension was filtered off, washed with acetic acid, then a mixture of acetic acid and water (1/2) and then water. Yield of 53% of a 87.9% pure product was obtained.
The above crude product was suspended in acetic acid and warmed to 105°C to 110°C until an orange brown solution is obtained. The solution was transferred to the crystallization vessel via a charcoal filter (or polish filtration) at a temperature above 85°C (optional step). The solution was then cooled to 80°C to 85°C. The mixture was stirred for one hour at 70°C to 80°C (optionally at 75°C) during which crystallization occurred. The product suspension was cooled to 20°C to 25°C for 17 hours or stirred for at least 12h at 20°C to 25 °C. The product suspension was filtered and washed with acetic acid, then acetic acid/ water (1/2) and finally water. The product was dried under vacuum at 50°C to 55°C. The yield of 70% corresponds to an overall yield of 44% for both parts of this preparation. The purity of the product assayed at 99.7% .
The preceding crystallization step is optional and the solution may be transferred to the crystallization vessel via polish filtration instead of via a charcoal filter.
The post crystallization suspension may be stirred for at least 12 hours at 20° C to 25 °C as an alternative to 17 hours.
Example 1b
Preparation of 4-hydroxy-5-methoxy-3 nitrobenzoic acid
A reactor was charged with 525 kg of glacial acetic acid and 50 kg vanillic acid. The mixture was heated with warm water gradually to 50°C in around 75 minutes. Temperature was set to 16°C. Nitric acid, 31.4 kg was then added gradually over a period of 3 hrs. When the administration was complete the mixture was allowed to stir for additional 3.5-4.5 hours.
The suspension was centrifuged whilst washed with 25 kg of acetic acid, 50 liter deionised water and 25 kg of acetic acid again. The wet crystalline material was suspended in 165 kg of acetic acid and heated at 91°C until complete dissolution. The solution was then cooled to 19.8°C and the mixture was allowed to stir for 1 hr. Centrifugation and washing with 15.2 kg acetic and 40 liter of deionised water was performed. The wet material was then dried in tray vacuum drier between 40-50°C until constant weight, for 72 hours. The dry material weight was 28.7 kg. The calculated yield was 45.4%.
Example 1c
Preparation of 4-h droxy-5-methoxy-3 nitrobenzoic acid

A suspension of vanillic acid (68.8 kg) in acetic acid (720 kg) is cooled to 17°C before an excess of a 65% nitric acid (44.0 kg) is added. After complete dosage of nitric acid the suspension is stirred for 2 hours. The suspension is filtered off and the wet cake is successively washed with acetic acid (80.0 kg), acetic acid/water (1:2 w/w – 105 kg) and finally water (80 kg – if necessary repeat). The solid is dried at 52°C for NMT 12 hours prior going to next step.
A suspension of the crude solid (650 kg) in acetic acid is warmed to 105 °C and stirred until complete dissolution of the crude solid. After polish filtration, the solution is cooled to 20°C over 3h resulting in crystallization and the suspension is stirred for 2h at 20°C. The solid is filtered off and the wet cake is successively washed with acetic acid (80 kg), acetic acid/water (1:2 w/w – 105 kg) and finally water (193 kg – if necessary repeat). 4-hydroxy-5-methoxy-3 nitrobenzoic acid pure is dried under vacuum at max. 55 °C until max 0.5% w/w residual acetic acid and max 0.2% w/w water is reached.
Example 2a
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoic acid
The process of Example la was scaled up to employ vanillic acid (375g) in acetic acid (3940g) to which was added nitric acid (65%, 245g) at 12°C over 3 hours followed by stirring for one hour. The overall yield was 40% of a 99.9% pure product.
Example 2b
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoic acid

Vanillic acid methyl ester (33g) and sodium nitrite (0.625g) are charged. Water (158mL) and 1,4-dioxane (158mL) are added at room temperature. The reaction mixture is heated to 40 °C. Nitric acid (65%) (15.75g) is added in the course of three hours and the resulting mixture is stirred for 4h after addition. The reaction mixture is sampled for completion.
The water/nitric-acid/dioxane azeotrope is distilled off in vacuum at 40 °C. The resulting product suspension is quenched by addition of sodium hydroxide solution (50% , 33.2 mL) and then stirred for 16h. The quench mixture is sampled for completion.
Then, HCl (18,5%, 70.2mL.) is added until the pH is below 1. The product is filtered off and washed with water (27.9mL). The product is then dried in vacuum at 50 °C. The overall yield was 81 % of a 97.3 % pure product.
Example 3a
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoyl chloride

A suspension of compound of Example la (1.0 eq) in dioxane (approx 4.5 vol) was treated with thionyl chloride (1.5 eq) and heated to 80°C. A clear solution formed at approximately 75 °C. The mixture was stirred for 3 hours at 80°C. Unreacted thionyl chloride was distilled off and after distillation the residue was cooled to 10°C.
Example 3 b
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoyl chloride
A suspension of compound of Example la (1.0 eq) in DCM (approx 3.4 vol) is treated with thionyl chloride (1.0 – 1.2 eq, for example 1.1 eq) and catalytic amount (0.011 eq) of DMF and the mixture is stirred for 16 h at 40°C. DCM is distilled off (approx 2.7 vol) and the residue is diluted with THF (approx 1.8 vol). The excess of thionylchloride is distilled off with THF/DCM and the residue after distillation is cooled to 10°C.
Example 3c
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoyl chloride
A suspension of compound of Example la (1.0 eq) in DCM (approx 4.5 vol) is treated with thionyl chloride (1.0 – 1.2 eq, for example 1.1 eq) and catalytic amount (0.0055 eq) of DMF and the mixture is stirred for 16 h at reflux. Unreacted thionylchloride is distilled off with DCM and the residue after distillation is diluted with THF (approx 1.8 vol) and cooled to 10°C.
The amount of DCM may be approx 3.4 as an alternative to approx 4.5 vol.
The catalytic amount of DMF may be about 0.011 eq as an alternative to 0.0055 eq.
Example 3d
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoyl chloride
In a reactor 68 kg dichloromethane, 20 kg 5-nitro- vanillic acid of example 1b, 76 gram of N,N-dimethylformamide and 13.4 kg (8 L) thionyl chloride, was charged at 20.2°C.
The mixture was heated to 40°C until all the starting material dissolved and the evolution of HCl and SO2 stopped. When all the starting material was consumed 5-10 L dichloromethane was distilled off at normal pressure at 40°C then the mixture was cooled to 20-25 °C and the distillation was continued until dry under vacuum at 40°C.
The evaporation residue was dissolved in 36 kg dry THF. The THF solution was used in
Example 4d.
Example 3e
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoyl chloride
A suspension of product of example 1C (4-hydroxy-5-methoxy-3 nitrobenzoic acid -160g, 1eq) in 1,4-dioxane (720mL, 4.5vol) is treated with thionyl chloride (169.8g, 103.7mL,1.5eq) and heated to 80°C. A clear solution is formed at approx. 75 °C. The mixture is stirred at 80 °C (3 hours). Unreacted thionyl chloride is distilled off and the residue after distillation is cooled to 10°C.
Example 4a
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
In this example the compound of formula (IV) is reacted with the compound of formula (V) to produce the compound of the formula (III).



Compound of formula (V) (1.24 eq) was suspended in 1,4-dioxane (approximately 4.5 vol) and the suspension cooled to 10°C. The acyl chloride (compound of formula (IV)) solution of Example 3a in 1,4-dioxane was added slowly maintaining the temperature below 20°C. A clear orange solution was formed. After complete addition, the reaction mixture was stirred at 20°C for one hour. Pyridine (approximately 8eq) was added and the reaction mixture heated slowly to 115°C. The mixture was stirred for 6 hours at 115°C and then cooled to 20°C.
The dioxane/pyridine was distilled off under vacuum at 70°C. The residue was kept at 80°C and ethanol (approx 8 vol) added to induce crystallization. The resulting yellow suspension was cooled to 0°C and stirred for two hours. The product was filtered off and washed with ethanol (2.5 vol) water (3.8 vol) and ethanol 2.5 vol). The product was dried under vacuum at 50 °C. Typical yields for this process are 82 to 85%.
In an optional variant, methanol was employed in place of ethanol to induce crystallization.
Example 4b
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
In a different reactor, compound of formula (V) (1.1 eq) is dissolved in DM Ac (approx 5.8 vol) and the solution is cooled to 5°C. The benzoyl chloride solution of Example 3b in THF/DCM is then added slowly maintaining the temperature below 10°C. After complete addition, the reaction mixture is stirred at 20 ±5°C. Pyridine (1.3 to 1.6 eq, for example 1.5 eq) is charged and the reaction mixture is heated slowly to 110±5°C removing low boiling components by distillation. The mixture is stirred for additional 3 h at 110±5°C.
In a further reactor, concentrated HCl (23.8 eq) is diluted with water (approx. 8.5 vol) and cooled to 10 °C. The reaction mixture in pyridine is dosed slowly to diluted hydrochloric acid. After complete addition, the resulting suspension is stirred for additional 2 h and the solid is filtered off. The crude solid is washed once with water and pre-dried on funnel.
The crude solid is suspended in DCM (approx. 28.6 vol) and the suspension is heated to 40°C to reach a clear solution. Resulting solution is cooled to 20°C and extracted with water. After phase separation, the aqueous phase is re-extracted with DCM and combined organic phase are washed once with water. DCM is distilled off under vacuum followed by addition of ethanol. Resulting suspension is further distilled to reduce the amount of DCM, then cooled to 5°C and stirred for additional 2 h. Finally, the product is filtered off, washed once with cold ethanol and dried under vacuum at 45°C.
Example 4c
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
In a second reactor, compound of formula (V) (1.1 eq) is dissolved in DMAc (approx. 7 vol) and the solution is cooled to 5°C. The benzoyl chloride solution of Example 3c in THF/DCM is added slowly maintaining the temperature below 10 °C. After complete addition, the reaction mixture is stirred at 20 ± 5°C for 30 min. Pyridine (6.9 to 7.3 eq, for example 7.14 eq) is charged and the reaction mixture is heated slowly to 110°C removing low boiling components by distillation. The mixture is stirred for additional 4 h at 110°C and cooled to 20°C.
In a third reactor an emulsion of diluted hydrochloric acid (prepared from cone. HCl (19.6 eq) and approx. 7.6 vol distilled water) and DCM (approx. 25.5 vol) is cooled to about 15 °C before the reaction mixture in pyridine is dosed slowly to the emulsion. After complete addition, the organic phase is separated and washed with water before DCM is distilled off under vacuum followed by addition of ethanol. The resulting suspension is further distilled to reduce the amount of DCM, then cooled to 5°C and stirred for additional 2 h.
Finally, the product is filtered off, washed once with cold ethanol and dried under vacuum at 45 °C.
Example 4d
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
140 kg Ν,Ν-dimethyl acetamide was charged into the reactor. 24.2 kg of amidoxime of Preparation 4 was dissolved in N,N-dimethyl acetamide while stirring at 21°C. The solution was cooled to 5-10°C. The THF solution of Example 3d was introduced slowly into the reaction mixture, 1.5-2 hrs, while the internal temperature was maintained at max. 9.5°C by external cooling. When the addition was complete the external cooling
was stopped. The internal temperature was allowed to raise to 21 °C in an hour. After stirring for 30 minutes, pyridine 53.0 kg was added to the mixture, while the temperature was in the range of 22.4°C – 20.6°C. Heating was started and the internal temperature raised to 105-115°C. The mixture started to reflux for 3h while the internal temperature managed to 113°C by partial distillation of some THF. The reaction mixture was then cooled and introduced to a mixture of 220 kg concentrated HCl and 170 kg of deionised water while the internal temperature was maintained between 14-16°C. The reactor was rinsed with 10 kg of Ν,Ν-dimethylacetamide and 20 kg deionised water. The rinse liquid was run to the mixture. The suspension was then further cooled to 5-10°C and stirred for 1.5-2.0 hours. The product was centrifuged and was washed 80 kg deionised water. Crude wet weight of the product was 88.6 kg.
The crude wet product, was dissolved in 460 kg (340 L) dichloromethane at max 40°C. When dissolved the temperature was set to 20-30°C and 120 kg deionised water was added. The organic phase was separated, the inorganic phase was extracted with 80 kg dichloromethane. The organic phase of 460 kg, was then washed with 200 kg deionised water and the phases were separated. The inorganic phase was extracted with the 80 kg dichloromethane and the organic phases were unified. The organic phase obtained so was concentrated in vacuum at 35°C to 200-240 Liter, then 260 kg ethanol 96% was continuously added and the evaporation was continued to a final 200-240 liter volume. Then the mixture was cooled to 5-10°C and was allowed to stir for 3 hrs. Centrifuging, washing with 20 kg ethanol resulted in 35.4 kg wet product. Vacuum drying for 16 hours at 45°C gave 34.09 kg dry product. The yield was 79.9%.
Example 4e
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
In a second vessel, (Z)-2,5-dichloro-N’-hydroxy-4,6-dimethylnicotiriimidamide (201.2g, 1.24eq) is suspended in 1,4-dioxane (720mL, 4.5vol) and the suspension is cooled to 10°C. The residue of example 3e in 1,4-dioxane is added slowly maintaining the temperature below 20°C. A clear orange solution is formed. After complete addition, the reaction mixture is stirred at 20°C for 1 hour. Pyridine (483.7mL, 8eq) is then charged and the reaction mixture is heated slowly to 115°C. The mixture is stirred at 115°C for 6 hours. The solution is then cooled to 20°C. Dioxane/pyridine is distilled off.
After distillation, the pit is kept at 80 °C and ethanol (1.28L, 8vol) is added at this temperature to induce crystallization. The resulting yellow suspension is cooled to 75 °C and stirred for 1h at this temperature to allow crystal growth. The product suspension is then cooled to 0 °C and stirred for 2h at this temperature. The product is filtered off and washed subsequently with ethanol (400mL, 2.5vol), water (608mL, 3.8vol) and ethanol (400mL, 2.5vol). The product is dried under vacuum at 50°C until LOD is max 1% w/w.
Example 4f
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
A mixture of compound of formula (V) (11.7g, 50 mmol, 1.25eq), methyl 4-hydroxy-3-methoxy-5-nitrobenzoate (10g, 40 mmol, leq) and a catalytic amount of p-toluenesulfonic acid (0.76g, 4mmol, 0.1eq) in dimethyl acetamide was heated to 80°C. The reaction was followed by HPLC. After 23h, 6% of conversion was obtained.
Example 4g
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-y1]-2-hydroxy-3-methoxy-1-nitrobenzene
A mixture of compound of formula (V) (11.7g, 50 mmol, 1.25eq), methyl 4-hydroxy-3-methoxy-5-nitrobenzoate (10g, 40 mmol, 1eq) and a catalytic amount of aluminum chloride (0.53g, 4mmol, 0.1eq) in dimethyl acetamide was heated to 80°C. The reaction was followed by HPLC. After 20h, 10% of conversion was obtained.
Example 5a
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene

A solution of the product of Example 4a (24g) was dissolved in dichloromethane (388g) at 20-40°C. The yellow solution was cooled to 5°C and urea hydrogen-peroxide (UHP) (17.6g) and trifluoroacetic acid anhydride (37g) added and stirring continued for 12hr at 5°C. The reaction mixture was warmed to room temperature over one hour and stirring continued for a further five hours. The precipitate that formed was filtered off and washed with dichloromethane. The combined filtrates were diluted further with dichloromethane, all washed and concentrated at atmospheric pressure. Toluene was added and the resulting suspension concentrated under vacuum, to remove residual dichloromethane. Further toluene was added and the mixture checked to ensure less than 0.5% dichloromethane and less than 0.1% water was present. Formic acid was added to provide a 10-12% formic acid in toluene mixture. The resulting suspension was warmed to 90°C and stirred until complete dissolution of solid. Crude product was obtained by cooling the solution to 5-10°C until crystallization commenced. The suspension was agitated at 5-10°C until crystallization appeared complete. The solid was filtered off, washed with toluene and dried under a stream of nitrogen.
The crude product was suspended in 10-12% wt/wt solution of formic acid in toluene and warmed to 90°C until dissolution of the solid. The solution was cooled to 5°C and stirred at 5°C until crystallisation occurred. The solid was obtained by filtration and washed with toluene. This recrystallization was repeated until the product tested as containing less than 0.1 % of starting material. The pure product was dried under vacuum at 50°C.
Example 5b
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
After dissolution of the product of Example 4b (24g) in DCM (388g) at 20-40°C the yellow solution is cooled to 5°C before the temperature controlled addition of urea hydrogen peroxide complex (UHP)(17.6) and trifluoroacetic anhydride (TFAA) (37g). After addition of TFAA is complete stirring is continued for 12h at 5°C before the reaction mixture is warmed to room temperature (RT) within 1 h and stirring is continued for additional 5 h. The precipitate formed during the reaction is filtered and washed with DCM on the funnel filter. The combined filtrates are diluted with DCM (325g) and then repeatedly washed with water before concentrated at atmospheric pressure. DCM is replaced by toluene (170g) and the resulting suspension is concentrated again under vacuum to remove surplus DCM. Distillates are replaced by fresh toluene as before and the mixture is analyzed for residual water and DCM (Residual DCM after solvent switch max. 0.5%; residual water after solvent switch max. 0.1 %). Formic acid (24g) is charged resulting in an approx. 10-12 % w/w formic acid in toluene solvent mixture The resulting suspension is warmed to 90°C and stirred until compete dissolution of the solid is achieved. The crude product is crystallized by cooling of this solution to 5-10°C and subsequent agitation of the resulting suspension at 5-10°C. The solid is filtered of washed with toluene and then dried in a stream of nitrogen gas.
The crude product so obtained is suspended in an approx. 10-12 %w/w solution (176g) of formic acid in toluene. The suspension is warmed to 90°C and stirred until all product is dissolved. After cooling of this solution to 5°C and subsequent stirring at 5°C, crude product is isolated by filtration and subsequent washing of the wet product with toluene.
The re-crystallization of crude product is repeated (2 or more times). The pure product (11.8g) is dried at 50°C under vacuum.
Example 5c
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
After dissolution of the product of Example 4c (24g) in DCM (388g) at 20-40°C the yellow solution is cooled to 5°C prior to the temperature controlled addition of urea hydrogen peroxide complex (UHP) (17.6g) and trifluoroacetic acid anhydride (TFAA) (37g). After addition of TFAA is complete stirring is continued for 12h at 5°C before the reaction mixture is warmed to RT within 1 h and stirring is continued for additional 5 h. The precipitate formed during the reaction is filtered and the filter cake is washed with DCM. The combined filtrates are diluted with DCM (325g) and then repeatedly washed with water before concentrated at atmospheric pressure. DCM is replaced by toluene (170g) and the resulting suspension is concentrated again in vacuum in order to remove surplus DCM and water. Distillates are replaced by fresh toluene followed by addition of formic acid (24g). The resulting suspension is warmed to 80°C and stirring is continued in order to dissolve the solid. The product is crystallized by cooling of this solution to 5°C and subsequent agitation of the resulting suspension at 5°C. The solid is filtered, washed with toluene and then dried in a stream of nitrogen gas.
The product is suspended in a formic acid / toluene (18g/158g) mixture followed by warming of the reaction mixture to 80°C. After dissolution of the product the solution is cooled to 5°C whereby the product precipitates. After additional stirring at 5°C the suspension is filtered and the filter cake is washed with toluene.
The re-crystallization of the product is repeated. The product is used as a wet material in the next process step (12.1g product obtained if dried at max. 60°C).
Example 5d
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yI]-2-hydroxy-3-mefhoxy-1-nitrobenzene
550 kg (420 L) Dichloromethane was charged into a reactor. 34 kg of product of example 4d was added to in a short period at 20°C internal temperature. The solution was cooled to 6.5°C then 24.9 kg urea hydrogen peroxide complex (UHP) was added over a period of 20-40 minutes between 5-10°C. Stirring was continued for additional 20 minutes between 6.5-7.5°C. Trifluoroacetic anhydride, 53 kg, was administered into the reaction mixture, starting and maintaining the temperature at 6-7°C over a period of 2-3 hours. When the administration was complete the mixture was stirred for additional 30 minutes. Then the internal temperature was allowed to rise to a maximum of 25°C over a period of 1.5 hours. The internal temperature was maintained between 20-25°C and the mixture was allowed to react for additional 18-20 hrs. The reaction mixture was centrifuged and the fuge was washed with 45 kg dichloromethane. To the separated dichloromethane solution 460 kg (350 L) dichloromethane and 190 kg deionised water was added. The mixture was stirred for 10 minutes and the phases were separated for 30 minutes. The organic phase was washed again with 2×190 kg deionised water and separated as previously. Evaporation of the unified organic solution at max 35 °C under vacuum was done to a final volume of 100-120 L. Then a total of 105 kg acetonitrile was administered into the system while the distillation was continued to keep the volume at 100-120 L. When complete an additional 170 kg (220 L) acetonitrile was added to the mixture at normal pressure. This suspension was heated to 70-80°C at normal pressure while dichloromethane was distilled off continuously. The mixture was then kept stirred for an hour. The suspension was cooled to 20-25°C and was stirred for an additional 30 minutes. The suspension was then centrifuged and was washed with 30 kg acetonitrile. The wet material, 29.7 kg, was vacuum dried for 16 hrs at 30°C. Dried product yield was 81.5%.
27.7 kg product, 240 kg toluene and 29.2 kg formic acid was charged into reactor then heated to 90°C for complete dissolution for 1 hour. Then the solution was cooled to 7°C and then the suspension was kept at 7°C for additional 2 hrs. If necessary seeding was applied with 3-5 grams of pure product. The suspension was then centrifuged for 1 hour whilst washing with 28 kg cold toluene. The product was suspended in 225 kg toluene and 27.2 kg formic acid was charged. The mixture then was heated to 90°C for complete dissolution for 1 hour. Then the solution was cooled to 20-25 °C, then the suspension was kept between 15-25°C for additional 2 hrs, seeded if necessary. The suspension then was centrifuged for 60 minutes whilst washed with 28 kg cold toluene. The recrystallization process may be repeated 2-3 more times.
Drying for 24 hrs at 38-41°C under vacuum was conducted until constant weight. This resulted in 16.34 kg (58.8%) dry material.
Example 5e
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
After dissolution of the product of Example 4e (150g) in DCM (2.43kg) at reflux, the yellow solution is cooled to 5°C prior to the temperature controlled addition of carbamide peroxide (UHP – urea hydrogen peroxide) ( 110g) and trifluoroacetic acid anhydride (TFAA) (155.1 ml in 4 portions within 2 hours). The mixture is stirred for 12h at 5°C then the reaction mixture is warmed to 25 °C over 1.5 hours and stirred for 5 hours. The precipitate formed during the reaction is filtered and the filter cake is washed with DCM (0.36 kg). The combined filtrates are warmed to 30°C and diluted with water (300g). 10% sodium hydroxide is added until pH= 4 is reached. The biphasic system is stirred for 10 minutes at 30°C and the mixture is then allowed to separate. The organic layer is then successively washed with a mixture water (750g) and 10% sodium hydroxide (7.5g) (until pH=4), 3.2% HCl solution (300g). DCM is distilled at atmospheric pressure and then replaced by toluene (1035g) applying vacuum (150mbar) and keeping internal temperature at 45°C. Formic acid (300g) and toluene (900g) are added keeping the internal temperature above 40°C. The resulting solution is distilled under vacuum (150 mbar, 45°C internal temperature) until distillation ceases. After seeding at 45°C, the slurry is stirred for 1 hour at 45°C then is cooled to 5°C over 2 hours. The suspension is stirred for at least 2 hours at 5°C and then filtered. The wet cake is washed with toluene (195g) and dried in a stream of nitrogen gas (Chemical purity of crude product min. 92 % area).
A suspension of crude product in formic acid (388g, 2wt) is warmed to 55°C and stirred until complete dissolution of the crude product. Toluene (1242g, 6.4wt) is added maintaining the internal temperature above 50 °C. The reaction is stirred at 150mBar and internal temperature 45 °C until distillation ceases. The vacuum and distillation is stopped and then seed is added at 45°C. The slurry is stirred for 1 hour at 45°C and cooled to 5°C in 2 hours. The resulting suspension is stirred for at least 2 hours at 5°C then filtered. The wet cake is washed with toluene (260g, 1.34wt). The wet cake is collected and charged into the reactor. This procedure is repeated at least twice until 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-y1]-2-hydroxy-3-methoxy-1-nitrobenzene level max is 0.1 % (a/a) prior to dry at 25°C max under vacuum.
Example 6
Example 5a was repeated on a larger scale employing product of Example 3 (82g), dichloromethane (1325g), urea peroxide (60.1g) and trifuoroacetic acid anhydride
(128g).
Example 7a
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol.

(Π)
Product of Example 6 (15g) was suspended in N-methyl pyrrolidone (NMP) (131.5g) and cooled to 5°C. Aluminium chloride (6.2g) and pyridine (12g) were added while maintaining the temperature at 5°C. After the addition of pyridine was complete the reaction mixture was warmed to 60 °C and maintained for 2 hours. After confirmation that less than 0.5 starting material remained, the reaction mixture was cooled, and aqueous HCl (water 233g, HCl 123g, 37%) added. The resulting yellow solid was isolated by suction filtration. The resulting wet product was washed with water and propan-2-ol (67g) and dried under vacuum.
Optionally, the crude product was suspended in ethanol (492g) and warmed to reflux. The suspension was stirred for 1 hour under reflux and then cooled to room temperature. The solid was obtained by filtration, washed with ethanol and dried under vacuum at 50°C. A typical yield of 85% was achieved.
If desired either the final ethanol crystallised material or the initially produced product after washing with propan-2-ol may be employed in preparation of micronized material for use in pharmaceutical compositions.
Example 7b
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-y1)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol.
An approx. 11 % w/w suspension of the product of example 5b (20g) in NMP (150g) is cooled to 5°C followed by a consecutive temperature controlled addition of aluminium chloride (8g) and pyridine (15.3g). After addition of pyridine is complete the reaction mixture is warmed to 60°C followed by additional 2 h reaction time. After complete conversion of the product of example 5b the reaction mixture is cooled before an aqueous diluted hydrochloric acid (water 293g, HCl 177g, 34%) is dosed. By addition of the hydrochloric acid, crude product precipitates from the NMP/water matrix as a yellow solid which is isolated by suction filtration. The resulting wet product is washed with water and 2-propanol in a replacement wash followed by drying of the wet crude product under vacuum.
The crude product is suspended in ethanol (282g) followed by warming of the mixture to reflux. The suspension is stirred for 1 h at reflux conditions followed by cooling to room temperature. The product is isolated by filtration of the suspension. The wet product is washed with ethanol and subsequently dried in vacuo at approx 50°C (typically weight corrected yield was 85%).
The product (20g) is suspended in formic acid (725g) before the resulting suspension is warmed to max. 67°C. Stirring is continued until complete dissolution of the product is achieved. The hot solution is filtered and the filtrate is cooled to 40 – 45°C before the product is precipitated first by concentration of the solution to approx. 40% (v/v) of its original volume followed by addition of the anti solvent 2-propanol (390g). After addition of 2-propanol is finished the resulting suspension is kept at 55-60°C for crystal ripening followed by cooling to RT and filtration. The filter cake is washed with 2-propanol followed by drying of the material at max. 58°C until loss on drying (LOD) max. 0.5% . Typically, a yield of 97-98% was obtained.
If desired the product may be employed in preparation of micronized material for use in pharmaceutical compositions.
Example 7c
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol.
A suspension of the product of example 5c (20g) or of example 6 (20g) in NMP (153g) is cooled to 5°C followed by a consecutive temperature controlled addition of aluminium chloride (8.2g) and pyridine (15.4g). After addition of pyridine is complete the reaction mixture is warmed to 60°C followed by additional 3 h reaction time. After complete conversion of the product of example 5c or of example 6 the crude product is
precipitated by a temperature controlled addition of an aqueous hydrochloric acid solution (water 296g, HCl 179g, 34%). Filtration of the solid followed by washing of the wet filter cake with water and 2-propanol yields a crude product wet material which is immediately dissolved in formic acid (536g). After polish filtration the filtrate is concentrated under vacuum followed by addition of the anti-solvent 2-propanol (318g). After aging of the resulting suspension at 55-60°C the suspension is cooled to RT and filtered. The wet filter cake is washed with 2-propanol. The wet product is dried under vacuum at max. 58°C until LOD max. 0.5%. The yield was in the range of 70-95%
If desired the product may be employed in preparation of micronized material for use in pharmaceutical compositions.
Example 7d
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol.
132 kg (147 L) N-methylpyrrolidone was charged into a 1000 L reactor. 16.3 kg of product of example 5d was then added. The suspension was cooled to 5-7°C and 6.5 kg of sublimed aluminium chloride was added in portions keeping the internal temperature between 5-10°C. The mixture was stirred for 10 minutes then 12.6 kg pyridine was added maintaining the internal temperature between 5-10°C. The mixture was warmed with water in the jacket to 20-25°C and the mixture was stirred for 30 minutes. Then the mixture is heated to 58-62 °C and reacted for around 2 hours. In a separate reactor a mixture of 240.5 kg deionised water and 146.4 kg concentrated HCl was mixed. This was cooled to 15-20°C. The reaction mixture from the demethylation was introduced into the diluted hydrochloric acid between 20-25°C. Optionally, 51.2 kg dichloromethane was added to the suspension, stirred for 30 minutes and was centrifuged, washed with 60 kg deionised water and 20 kg isopropanol. Drying gave 15.9 kg of product.
The product was suspended in 185.3 kg of ethanol. The mixture was then stirred at 78°C for an hour, then cooled to 20-25°C and stirred for 1 hour. The suspension was then centrifuged and the filtercake was washed with 44.5 kg ethanol, 96% . The solid material was dried at 50°C in vacuum in a stainless steel tray drier. 14.35 kg (90.3% yield) dry product was obtained.
A reactor was charged with 317.2 kg formic acid and dry product. The mixture was heated to 65 °C until all the solid dissolves. The hot solution was then filtered to an empty 1000 L reactor, was rinsed with 20 kg formic acid, then the formic acid solution was distilled partially off under vacuum to around 80-100L. 260 kg isopropanol was then introduced at 50-60°C and stirred for 30-35 minutes. The mixture was then cooled to 20-25°C with water in the jacket and was allowed to stir min 2 hours. The suspension was then centrifuged and was washed with 25 kg isopropanol. The wet material was removed from the fuge and was transferred into vacuum tray drier and was dried until constant weight under vacuum at 45-50°C resulting in 13.6 kg product, with a yield of 95.3% .
If desired the product may be employed in preparation of micronized material for use in pharmaceutical compositions.
Example 7e
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol.
A suspension of product of Example 5e (34.1kg) in N-Methyl pyrrolidone (NMP) (182kg) is warmed to 50 °C until dissolution and then cooled to 5°C followed by a consecutive temperature controlled addition of aluminium chloride (9.8 kg) and pyridine (18.2kg). After addition of pyridine is complete the reaction mixture is warmed to 60°C and stirred for at least 2 hours. The reaction mixture is cooled to 10-16°C (e.g. 11, 13, 15°C) before an aqueous diluted hydrochloric acid (4M solution, 283L) is dosed maintaining the temperature below 25 °C. During the addition of the hydrochloric acid the crude product is precipitated from the NMP/water matrix as a yellow solid. The yellow solid is filtered and subsequently washed with water (179kg), 2-propanol (105kg). The wet solid is dried under vacuum at 55°C.
A suspension of wet product (25.1kg) in formic acid (813kg) is warmed to max. 67°C. The mixture is stirred at 67°C until complete dissolution of the product is achieved. The hot solution is filtered and the filtrate is cooled to 40 – 45°C before the product is precipitated first by concentration of the solution to approx. 40% (v/v) of its original volume followed by addition of the anti solvent 2-propanol (380kg). After addition of 2-propanol the resulting suspension is stirred at 55-60°C for crystal ripening followed by cooling to RT and filtration. The filter cake is washed with 2-propanol (38kg) and then dried at max. 58°C until LOD max. 0.5%). The product may be milled (for example using the method of Example 8).
Example 8
Micronization of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol with MC JETMILL® type 200 milling equipment (micronization through spiral jet mills)
Equipment:
Mill: MC JETMILL® 200
Dosing unit: K-Tron T 35
Cyclone: type 600
Each micronization trial was performed on at least 2 kg of 5-(3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol.
The following working parameters have been defined for the micronization:
Feed rate range: 24.0-48.0 kg/h (200-400 g/30sec.)
Mill pressure range: 3.0-4.0 bar
Venturi pressure range: 3.0-4.0 bar; preferably the Venturi pressure is the same as the mill pressure
Using the above equipment and working parameters the microparticles of 5-(3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol comply with the following particle size specification (particle size determined by optical microscopy): D10 (EDC) is not less than 4 or 5 μm (for example not less than 5 μm), the D50 (EDC) is 10-45 or 15-30 μm (for example 15-30 μm) and the D95 (EDC) is not more than 60 or 70 μm (for example not more than 60 μm).
Example 9 (Figure 5)
2,5-Dichloro-4,6-dimethyl-nicotinonitrile is reacted with hydroxylamine in the presence of catalytic amounts of 1,10-phenanthroline monohydrate to yield the aldoxime (Z)-2,5-dichloro-N’-hydroxy-4,6-dimethylnicotinimidamide which represents the first coupling partner towards the synthesis of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene. The second coupling partner 5-nitro-vanillic acid pure is synthesized from vanillic acid by nitration with 65 % nitric acid followed by re-crystallization of the crude 5-nitro-vanillic acid intermediate from acetic acid. The convergent assembly of the oxadiazole moiety in 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene is achieved by first activation of 5-nitro-vanillic acid as its acid chloride and subsequent coupling with the aldoxime (Z)-2,5-dichloro-N’-hydroxy-4,6-dimethylnicotinimidamide. Cyclisation of the initially formed coupling product is achieved thermally to give the oxadiazole moiety by elimination of water. The reaction
mixture of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene, after ring closure reaction, is concentrated and product isolated from 1,4-dioxane/ethanol mixture in one step. Oxidation of the pyridine ring to the corresponding aryl-N-oxide (5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene) is achieved with trifluoroperoxoacetic acid which is formed in situ from UHP (Urea hydrogen peroxide complex) and trifluoroacetic acid anhydride. Unreacted 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene is subsequently removed from 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene by repeated re-crystallisation from formic acid/toluene. The analogue intermediate 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene pure with a level of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene below 0.10 %area is converted to 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol crude analogue by ether cleavage in the presence of a stoichiometric amount of aluminium chloride and pyridine. After completion of the reaction, the crude product is isolated by precipitation with an aqueous hydrochloric acid followed by dissolution of the precipitate in formic acid. After polish filtration of the resulting solution and partial solvent switch from formic acid to isopropanol, 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol is crystallized from the resulting formic acid/IPA crystallization matrix and finally optionally milled to the desired particle size.

PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012107708
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007117165
scheme 1 depicts example how to produce a compound of the general formula IIB from a compound of the general formula IVB:

iii.

HB IVB
Scheme 1. Reagents: i. Piperidine, ethanol, reflux; ii. SO2Cl2, CCl4, reflux; iii. POCI3, 120 0C, 18 h; iv. 50% H2NOH, MeOH-H20, 1.25 mol % 1,10-phenanthroline hydrate.
The following reaction scheme 2 depicts an example how to produce certain compounds of general formula III:

I, R8 = methyl III, R8 = R9 = H
iv.
R9 = H

III, R8 = R9 = benzyl
Scheme 2. Reagents: i. 65 % HNO3, AcOH; ii. 48 % HBr (aq), 140 0C; iii. MeOH, HCl(g); iv. BnBr, K2CO3, CH3CN, reflux; v. 3N NaOH, MeOH/H2O.
The following reaction scheme 3 depicts an example how to produce the compound A, by activation of a compound according to general formula III followed by cyclisation involving condensation with a compound according to formula HB, dehydration and deprotection of the methyl residue protecting the hydroxyl group;
0C

compound A
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2007013830A1 | Jul 26, 2006 | Feb 1, 2007 | Portela & Ca. S.A. | Nitrocatechol derivatives as comt inhibitors |
| WO2007117165A1 | Apr 10, 2007 | Oct 18, 2007 | Bial – Portela & Ca, S.A. | New pharmaceutical compounds |
| WO2008094053A1 * | Oct 10, 2007 | Aug 7, 2008 | Bial-Portela & Ca, S.A. | Dosage regimen for comt inhibitors |
| WO2012107708A1 * | Oct 21, 2011 | Aug 16, 2012 | Bial – Portela & Ca, S.A. | Administration regime for nitrocatechols |
| US20100168113 * | Apr 10, 2007 | Jul 1, 2010 | David Alexander Learmonth | Pharmaceutical Compounds |
| Reference | ||
|---|---|---|
| 1 | * | “[1,2,4]-oxadiazolyl nitrocatechol derivatives“, IP.COM JOURNAL, IP.COM INC., WEST HENRIETTA, NY, US, 3 May 2012 (2012-05-03), XP013150541, ISSN: 1533-0001 |
| 2 | * | KISS L E ET AL: “Discovery of a long-acting, peripherally selective inhibitor of catechol-O-methyltransferase“, JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, US, vol. 53, no. 8, 22 April 2010 (2010-04-22), pages 3396 – 3411, XP002594266, ISSN: 0022-2623, [retrieved on 20100324], DOI: 10.1021/JM1001524 |
| 3 | L. E. KISS ET AL., J. MED. CHEM., vol. 53, 2010, pages 3396 – 3411 | |
| 4 | * | RASENACK N ET AL: “MICRON-SIZE DRUG PARTICLES: COMMON AND NOVEL MICRONIZATION TECHNIQUES“, PHARMACEUTICAL DEVELOPMENT AND TECHNOLOGY, NEW YORK, NY, US, vol. 9, no. 1, 1 January 2004 (2004-01-01), pages 1 – 13, XP009055393, ISSN: 1083-7450, DOI: 10.1081/PDT-120027417 |
REFERENCES
1: Bicker J, Alves G, Fortuna A, Soares-da-Silva P, Falcão A. A new PAMPA model using an in-house brain lipid extract for screening the blood-brain barrier permeability of drug candidates. Int J Pharm. 2016 Jan 30. pii: S0378-5173(16)30072-2. doi: 10.1016/j.ijpharm.2016.01.074. [Epub ahead of print] PubMed PMID: 26836708.
2: Devos D, Moreau C. Opicapone for motor fluctuations in Parkinson’s disease. Lancet Neurol. 2015 Dec 22. pii: S1474-4422(15)00346-4. doi: 10.1016/S1474-4422(15)00346-4. [Epub ahead of print] PubMed PMID: 26725545.
3: Ferreira JJ, Lees A, Rocha JF, Poewe W, Rascol O, Soares-da-Silva P; Bi-Park 1 investigators. Opicapone as an adjunct to levodopa in patients with Parkinson’s disease and end-of-dose motor fluctuations: a randomised, double-blind, controlled trial. Lancet Neurol. 2015 Dec 22. pii: S1474-4422(15)00336-1. doi: 10.1016/S1474-4422(15)00336-1. [Epub ahead of print] PubMed PMID: 26725544.
4: Rascol O, Perez-Lloret S, Ferreira JJ. New treatments for levodopa-induced motor complications. Mov Disord. 2015 Sep 15;30(11):1451-60. doi: 10.1002/mds.26362. Epub 2015 Aug 21. Review. PubMed PMID: 26293004.
5: Gonçalves D, Alves G, Fortuna A, Soares-da-Silva P, Falcão A. Development of a liquid chromatography assay for the determination of opicapone and BIA 9-1079 in rat matrices. Biomed Chromatogr. 2016 Mar;30(3):312-22. doi: 10.1002/bmc.3550. Epub 2015 Aug 17. PubMed PMID: 26147707.
6: Ferreira JJ, Rocha JF, Falcão A, Santos A, Pinto R, Nunes T, Soares-da-Silva P. Effect of opicapone on levodopa pharmacokinetics, catechol-O-methyltransferase activity and motor fluctuations in patients with Parkinson’s disease. Eur J Neurol. 2015 May;22(5):815-25, e56. doi: 10.1111/ene.12666. Epub 2015 Feb 4. PubMed PMID: 25649051.
7: Bonifácio MJ, Torrão L, Loureiro AI, Palma PN, Wright LC, Soares-da-Silva P. Pharmacological profile of opicapone, a third-generation nitrocatechol catechol-O-methyl transferase inhibitor, in the rat. Br J Pharmacol. 2015 Apr;172(7):1739-52. doi: 10.1111/bph.13020. Epub 2015 Jan 20. PubMed PMID: 25409768; PubMed Central PMCID: PMC4376453.
8: Kiss LE, Soares-da-Silva P. Medicinal chemistry of catechol O-methyltransferase (COMT) inhibitors and their therapeutic utility. J Med Chem. 2014 Nov 13;57(21):8692-717. doi: 10.1021/jm500572b. Epub 2014 Sep 2. PubMed PMID: 25080080.
9: Rocha JF, Falcão A, Santos A, Pinto R, Lopes N, Nunes T, Wright LC, Vaz-da-Silva M, Soares-da-Silva P. Effect of opicapone and entacapone upon levodopa pharmacokinetics during three daily levodopa administrations. Eur J Clin Pharmacol. 2014 Sep;70(9):1059-71. doi: 10.1007/s00228-014-1701-2. Epub 2014 Jun 14. PubMed PMID: 24925090.
10: Rocha JF, Santos A, Falcão A, Lopes N, Nunes T, Pinto R, Soares-da-Silva P. Effect of moderate liver impairment on the pharmacokinetics of opicapone. Eur J Clin Pharmacol. 2014 Mar;70(3):279-86. doi: 10.1007/s00228-013-1602-9. Epub 2013 Nov 24. PubMed PMID: 24271646.
11: Bonifácio MJ, Sutcliffe JS, Torrão L, Wright LC, Soares-da-Silva P. Brain and peripheral pharmacokinetics of levodopa in the cynomolgus monkey following administration of opicapone, a third generation nitrocatechol COMT inhibitor. Neuropharmacology. 2014 Feb;77:334-41. doi: 10.1016/j.neuropharm.2013.10.014. Epub 2013 Oct 19. PubMed PMID: 24148813.
12: Gonçalves D, Alves G, Fortuna A, Soares-da-Silva P, Falcão A. An HPLC-DAD method for the simultaneous quantification of opicapone (BIA 9-1067) and its active metabolite in human plasma. Analyst. 2013 Apr 21;138(8):2463-9. doi: 10.1039/c3an36671e. PubMed PMID: 23476919.
13: Rocha JF, Almeida L, Falcão A, Palma PN, Loureiro AI, Pinto R, Bonifácio MJ, Wright LC, Nunes T, Soares-da-Silva P. Opicapone: a short lived and very long acting novel catechol-O-methyltransferase inhibitor following multiple dose administration in healthy subjects. Br J Clin Pharmacol. 2013 Nov;76(5):763-75. doi: 10.1111/bcp.12081. PubMed PMID: 23336248; PubMed Central PMCID: PMC3853535.
14: Almeida L, Rocha JF, Falcão A, Palma PN, Loureiro AI, Pinto R, Bonifácio MJ, Wright LC, Nunes T, Soares-da-Silva P. Pharmacokinetics, pharmacodynamics and tolerability of opicapone, a novel catechol-O-methyltransferase inhibitor, in healthy subjects: prediction of slow enzyme-inhibitor complex dissociation of a short-living and very long-acting inhibitor. Clin Pharmacokinet. 2013 Feb;52(2):139-51. doi: 10.1007/s40262-012-0024-7. PubMed PMID: 23248072.
15: Palma PN, Bonifácio MJ, Loureiro AI, Soares-da-Silva P. Computation of the binding affinities of catechol-O-methyltransferase inhibitors: multisubstate relative free energy calculations. J Comput Chem. 2012 Apr 5;33(9):970-86. doi: 10.1002/jcc.22926. Epub 2012 Jan 25. PubMed PMID: 22278964.
16: Gonçalves D, Alves G, Soares-da-Silva P, Falcão A. Bioanalytical chromatographic methods for the determination of catechol-O-methyltransferase inhibitors in rodents and human samples: a review. Anal Chim Acta. 2012 Jan 13;710:17-32. doi: 10.1016/j.aca.2011.10.026. Epub 2011 Oct 20. Review. PubMed PMID: 22123108.
17: Kiss LE, Ferreira HS, Torrão L, Bonifácio MJ, Palma PN, Soares-da-Silva P, Learmonth DA. Discovery of a long-acting, peripherally selective inhibitor of catechol-O-methyltransferase. J Med Chem. 2010 Apr 22;53(8):3396-411. doi: 10.1021/jm1001524. PubMed PMID: 20334432.
////////BIA-9-1067, ONO-2370, BIA-91067, 923287-50-7, Opicapone, Catechol-O-methyl transferase, COMT inhibitor, Parkinson’s disease, PD, BIA 9-1067, BIA 91067, BIA-91067, BIA91067, EU 2016
OC1=CC(C2=NC(C3=C(Cl)[N+]([O-])=C(C)C(Cl)=C3C)=NO2)=CC([N+]([O-])=O)=C1O