It's only fair to share...














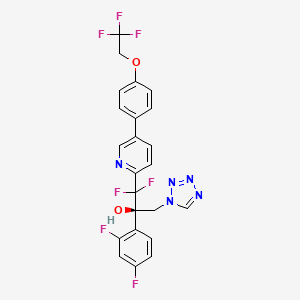
 OTESECONAZOLE
VT 1161
オテセコナゾール;
(2R)-2-(2,4-difluorophenyl)-1,1-difluoro-3-(tetrazol-1-yl)-1-[5-[4-(2,2,2-trifluoroethoxy)phenyl]pyridin-2-yl]propan-2-ol
OTESECONAZOLE
VT 1161
オテセコナゾール;
(2R)-2-(2,4-difluorophenyl)-1,1-difluoro-3-(tetrazol-1-yl)-1-[5-[4-(2,2,2-trifluoroethoxy)phenyl]pyridin-2-yl]propan-2-ol
C23H16F7N5O2
|
|
| Synonyms |
VT 1161 Oteseconazole
1340593-59-0
|
|---|
Other Names
- (αR)-α-(2,4-Difluorophenyl)-β,β-difluoro-α-(1H-tetrazol-1-ylmethyl)-5-[4-(2,2,2-trifluoroethoxy)phenyl]-2-pyridineethanol
- (2R)-2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-1,2,3,4-tetrazol-1-yl)- 1-{5-[4-(2,2,2-trifluoroethoxy)phenyl]pyridin-2-yl}propan-2-ol
Names
Oteseconazole is the international nonproprietary name (INN).[4] Oteseconazole is an azole antifungal used to prevent recurrent vulvovaginal candidiasis in females who are not of reproductive potential. Oteseconazole, also known as VT-1161, is a tetrazole antifungal agent potentially for the treatment of candidal vaginal infection. VT-1161 Protects Immunosuppressed Mice from Rhizopus arrhizus var. arrhizus Infection. VT-1161 dosed once daily or once weekly exhibits potent efficacy in treatment of dermatophytosis in a guinea pig model.
Oteseconazole has been used in trials studying the treatment of Tinea Pedis, Onychomycosis, Candidiasis, Vulvovaginal, and Recurrent Vulvovaginal Candidiasis.
Mycovia Pharmaceuticals is developing oteseconazole, the lead from a program of metalloenzyme Cyp51 (lanosterol demethylase) inhibitors, developed using the company’s Metallophile technology, for treating fungal infections including onychomycosis and recurrent vulvovaginal candidiasis (RVVC). In July 2021, oteseconazole was reported to be in phase 3 clinical development. Licensee Jiangsu Hengrui Medicine is developing otesaconazole, as an oral capsule formulation, for treating fungal conditions, including RVVC, onychomycosis and invasive fungal infections, in Greater China and planned for a phase 3 trial in April 2021 for treating VVC.
- OriginatorViamet Pharmaceuticals
- DeveloperMycovia Pharmaceuticals; Viamet Pharmaceuticals
- ClassAntifungals; Foot disorder therapies; Pyridines; Small molecules; Tetrazoles
- Mechanism of Action14-alpha demethylase inhibitors
- PreregistrationVulvovaginal candidiasis
- Phase IIOnychomycosis
- No development reportedTinea pedis
- 01 Jun 2021Preregistration for Vulvovaginal candidiasis (In adolescents, In adults, In children, Recurrent) in USA (PO)
- 01 Jun 2021Mycovia intends to launch otesaconazole (Recurrent) for Vulvovaginal candidiasis in the US in early 2022
- 06 Jan 2021Interim efficacy and adverse events data from a phase III ultraVIOLET trial in Vulvovaginal candidiasis released by Mycovia Pharmaceuticals
PATENT
WO 2017049080
WO 2016149486
US 20150024938
WO 2015143172
WO 2015143184
WO 2015143180
WO 2015143142
WO 2013110002
WO 2013109998
WO 2011133875
PATENT
WO 2017049080,
The function of metalloenzymes is highly dependent on the presence of metal ions in the active site of the enzyme. It is recognized that reagents that bind to and inactivate metal ions at the active site greatly reduce the activity of the enzyme. Nature uses this same strategy to reduce the activity of certain metalloenzymes during periods when enzyme activity is not needed. For example, the protein TIMP (tissue inhibitor of metalloproteinases) binds to zinc ions in the active sites of various matrix metalloproteinases, thereby inhibiting enzyme activity. The pharmaceutical industry has used the same strategy in the design of therapeutic agents. For example, the azole antifungal agents fluconazole and voriconazole contain 1-(1,2,4-triazole) group, which exists in the active site of the target enzyme lanosterol demethylase The heme iron binds, thereby inactivating the enzyme. Another example includes zinc-bound hydroxamic acid groups, which have been introduced into most of the published inhibitors of matrix metalloproteinases and histone deacetylases. Another example is the zinc-binding carboxylic acid group, which has been introduced into most of the published angiotensin converting enzyme inhibitors.
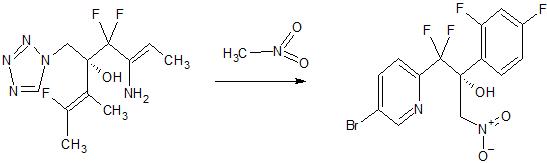
VT-1161, the compound 2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2, 2,2-Trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol, is an antifungal drug developed by VIAMET, currently in the clinical research stage, its structure is as follows Shown:
This compound mainly acts on the CYP51 target of fungal cells. Compared with the previous triazole antifungal drugs, it has the advantages of wider antibacterial spectrum, low toxicity, high safety and good selectivity. However, this compound is not suitable for Liquid preparations (including or excluding the parenteral delivery carrier) are used to treat patients in need thereof.
2-(2,4-Difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2,2-trifluoro Ethoxy)phenyl)pyridin-2-yl)propan-2-yl dihydrogen phosphate is a prodrug of VT-1161.
On the other hand, nearly half of the drug molecules are in the form of salts, and salt formation can improve certain undesirable physicochemical or biological properties of the drug. Relative to 2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2,2- Trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-yl dihydrogen phosphate, it is of great significance to develop salts with more excellent properties in terms of physical and chemical properties or pharmaceutical properties.
To this end, the present disclosure provides a new pharmaceutically acceptable salt form of a metalloenzyme inhibitor.
Example 1:
[0161]
(R)-2-(2,4-Difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2, 2-Trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-yl phosphate disodium salt (Compound 1)
[0162]
[0163]
(R)-2-(2,4-Difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2 ,2-Trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-yl phosphate (compound 1a, prepared according to the method of patent WO2013110002, 0.28g, 0.46mmol, 1.0eq) and ethanol (5mL ) Add to the reaction flask and stir evenly. A solution of NaOH (36.90 mg, 2.0 eq) dissolved in water (1 mL) was added dropwise into the above reaction flask, stirring was continued for 2 h, and concentrated to obtain compound 1, 300 mg of white solid.
[0164]
After X-ray powder diffraction detection, the XRPD spectrum has no sharp diffraction peaks, as shown in FIG. 10.
[0165]
Ms:608.10[M-2Na+3H] + .
[0166]
Ion chromatography detected that the sodium ion content was 6.23%.
[0167]
Example 2: (R)-((2-(2,4-Difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4 -(2,2,2-Trifluoroethoxy)phenyl)pyridin-2-yl)prop-2-yl)oxy)methyl phosphate disodium salt (compound 2)
[0169]
Under ice-cooling, NaH (58mg, 0.87mmol) was added to the reaction flask, 1.5mL of N,N-dimethylformamide and 0.6mL of tetrahydrofuran were added, followed by iodine (38mg, 0.15mmol), and then Compound 2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2,2-tri Fluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (2b, prepared according to the method of patent WO2013110002, 158mg, 0.3mmol) tetrahydrofuran (1ml) solution was added to the reaction solution, stirred and reacted for 1-4h , And then add compound 2a (519mg, 2.01mmol) in tetrahydrofuran (1ml) solvent to the reaction, stir until the reaction is complete, 10% aqueous ammonium chloride solution to quench the reaction, extract, concentrate and drain, the crude product 2c is directly used for the next One-step reaction, Ms: 750.0[M+H] + .
[0170]
[0171]
Under ice-bath cooling, add trifluoroacetic acid (0.5mL) to the crude product 2c (300mg) in dichloromethane (2mL) solution, stir until the reaction is complete, and after concentration, the target compound 2d, 82mg, Ms was separated by high performance liquid phase separation. :638.0[M+H] + .
[0172]
Add compound 2d (0.29g, 0.46mmol, 1.0eq) and ethanol (5mL) obtained in the previous step into the reaction flask, stir, and add NaOH (36.90mg, 2.0eq) water (1ml) solution dropwise to the aforementioned reaction solution , Stirred for 2-5 h, and concentrated to obtain 2,313 mg of the target compound.
Ms:638.10[M-2Na+3H] + .
EXAMPLE 11

2-(2,4-Difluorophenyl)-l,l-difluoro-3-(lH-tetrazol-l-yl)-l-(5-(4-(2,2,2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (11)
Compound 11 was prepared using the conditions employed for 1: 0.33 g as a solid. The precursor l-bromo-4-(2,2,2-trifluoroethoxy)benzene was prepared as described below in one step.
1H NMR (500 MHz, CDC13): δ 8.76 (s, 1 H), 8.70 (s, 1 H), 7.95 (d, / = 8.0 Hz, 1 H), 7.70 (s, 1 H), 7.64 (d, / = 8.5 Hz, 1 H), 7.54 (d, / = 8.5 Hz, 2 H), 7.42- 7.37 (m, 1 H), 7.08 (d, / = 8.5 Hz, 2 H), 6.79- 6.75 (m, 1 H), 6.69- 6.66 (m, 1 H), 5.58 (d, / = 14.0 Hz, 1 H), 5.14 (d, / = 14.0 Hz, 1 H), 4.44 – 4.39 (m, 2 H). HPLC: 99.1%. MS (ESI): m/z 528 [M++l].
Chiral preparative HPLC Specifications for (+)-ll:
Column: Chiralpak IA, 250 x 4.6mm, 5u
Mobile Phase: A) w-Hexane, B) IPA
Isocratic: A: B (65:35)
Flow Rte: l.OO mL/min
Optical rotation [a]D: + 24° (C = 0.1 % in MeOH). 1 -Bromo-4-( 2,2,2-trifluoroethoxy )benzene
To a stirred solution of trifluoroethyl tosylate (1.5 g, 5.8 mmol) in DMF (20 mL) was added K2CO3 (4 g, 29.4 mmol) followed by addition of p-bromo phenol (1.1 g, 6.46 mmol) at RT under inert atmosphere. The reaction mixture was stirred at 120 °C for 6 h. The volatiles were evaporated under reduced pressure; the residue was diluted with water (5 mL) and extracted with ethyl acetate (3 x 30 mL). The organic layer was washed with water, brine and dried over anhydrous Na2S04, filtered and concentrated in vacuo. The crude compound was purified by silica gel column chromatography eluting with 5% EtOAc/hexane to afford the desired product (0.8 g, 3.13 mmol, 53.3%) as semi solid. 1H NMR (200 MHz, CDC13): δ 7.44 – 7.38 (m, 2 H), 6.86-6.80 (m, 2 H), 4.38- 4.25 (m, 2 H).
Examples
The present invention will now be demonstrated using specific examples that are not to be construed as limiting.
General Experimental Procedures
Definitions of variables in the structures in schemes herein are commensurate with those of corresponding positions in the formulae delineated herein.
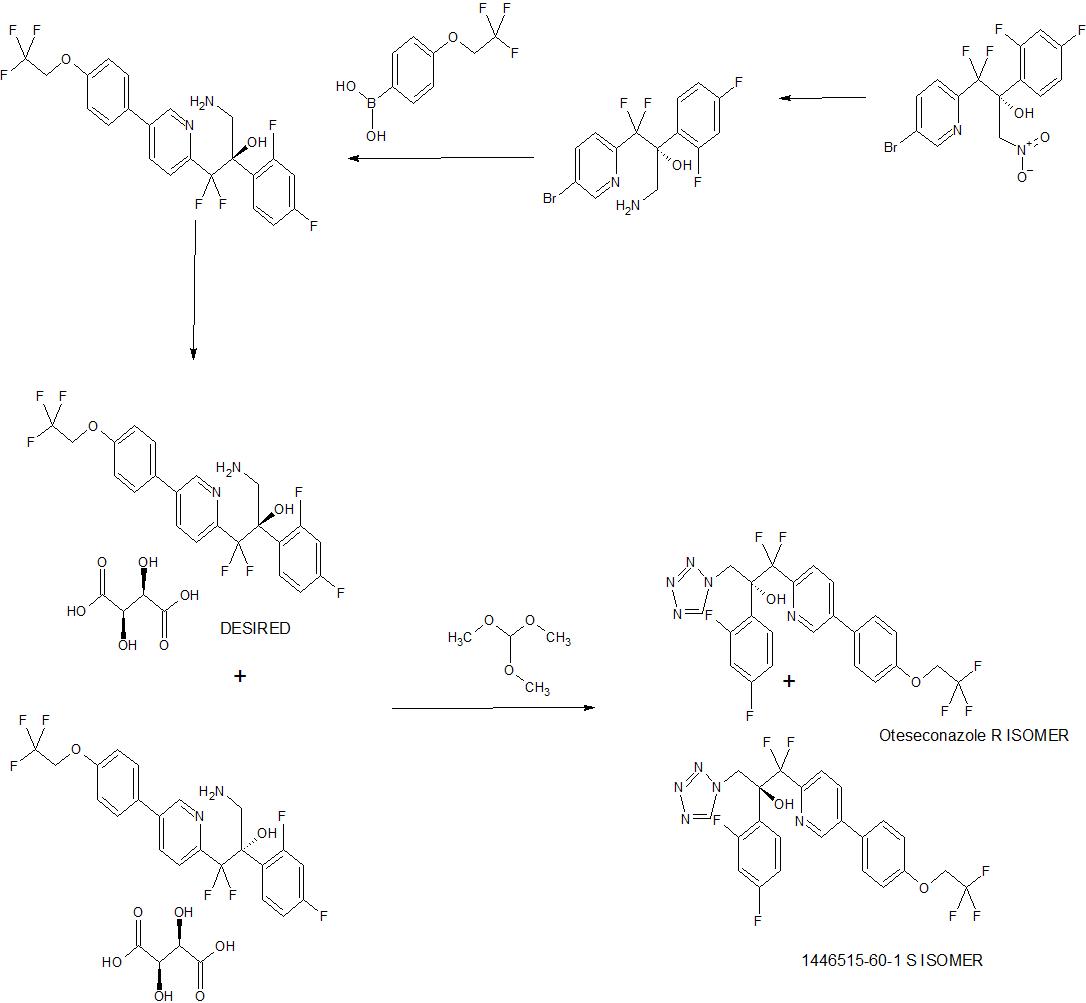
Synthesis of 1 or la

A process to prepare enantiopure compound 1 or la is disclosed. Syntheses of lor la may be accomplished using the example syntheses that are shown below (Schemes 1-4). The preparation of precursor ketone 3-Br is performed starting with reaction of 2,5-dibromo- pyridine with ethyl 2-bromo-difluoroacetate to produce ester 2-Br. This ester can be reacted with morpholine to furnish morpholine amide 2b-Br, followed by arylation to provide ketone 3-Br. Alternatively, ketone 3-Br can be afforded directly from ester 2-Br as shown in Scheme 1. Scheme 1. Synthesis of ketone 3-Br r

Ketone 3 may be prepared in an analogous fashion as described in Scheme 1 starting from corresponding substituted 2-bromo-pyridines, which can be prepared according to synthetic transformations known in the art and contained in the references cited herein (Scheme 2).
Scheme 2. Synthesis of ketone 3

R-i = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, – 0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, – 0(S02)-aryl, or -0(S02)-substituted aryl.
Alternatively, compound 1 can be prepared according to Scheme 3 utilizing diols 2-6b (or 2- 6d, the enantiomer of 2-6b, or mixtures thereof) or 2-6a (or 2-6c, the enantiomer of 2-6a, or mixtures thereof). Olefins 2-5a and 2-5 can be prepared by reacting ketones 3 and 1-4 under Wittig olefination conditions (e.g., Ph3PCH3Br and BuLi). Also, as indicated in Scheme 5, any of pyridine compounds, 3, 2-5a, 2-6b, 2-7b, 4*, 4b, or 6 can be converted to the corresponding 4-CF3CH2O-PI1 analogs (e.g., 1-4, 2-5, 2-6a, 2-7a, 5*, 1-6*, or 1 or the corresponding enantiomers, or mixtures thereof) by cross-coupling with 4,4,5, 5-tetramethyl-2- (4-(2,2,2-trifluoroethoxy)phenyl)-l,3,2-dioxaborolane (or the corresponding alkyl boronates or boronic acid or the like), in a suitable solvent system (e.g., an organic-aqueous solvent mixture), in the presence of a transition metal catalyst (e.g., (dppf)PdCl2), and in the presence of a base (e.g., KHCO3, K2C03, Cs2C03, or Na2C03, or the like). Olefins 2-5a and 2-5 can be transformed to the corresponding chiral diols, 2-6b (or 2-6d, the enantiomer of 2-6b, or mixtures thereof) or 2-6a (or 2-6c, the enantiomer of 2-6a, or mixtures thereof), through exposure to Sharpless asymmetric dihydroxylation conditions: 1) commercially available AD- mix alpha or AD-mix beta with or without additional osmium oxidant and methanesulfonamide, 2) combination of a catalytic osmium oxidant (e.g., Os04 or K20sC>2(OH)4), a stoichiometric iron oxidant (e.g., K3Fe(CN)6), a base (e.g., KHCO3, K2CO3, Cs2C03, or Na2C03, or the like), and a chiral ligand (e.g., (DHQ)2PHAL, (DHQD)2PHAL, (DHQD)2AQN, (DHQ)2AQN, (DHQD)2PYR, or (DHQ)2PYR; preferably (DHQ)2PHAL, (DHQD)2PHAL, (DHQD)2AQN, and (DHQD)2PYR), or 3) option 2) with methanesulfonamide. The primary alcohol of the resultant chiral diols, 2-6b (or 2-6d, the enantiomer of 2-6b, or mixtures thereof) or 2-6a (or 2-6c, the enantiomer of 2-6a, or mixtures thereof), can then be activated to afford compounds 2-7b (or 2-7d, the enantiomer of 2-7b, or mixtures thereof) or 2-7a (or 2-7c, the enantiomer of 2-7a, or mixtures thereof). For example, the mesylates can be prepared by exposing chiral diols, 2-6b (or 2-6d, the enantiomer of 2-6b, or mixtures thereof) or 2-6a (or 2-6c, the enantiomer of 2-6a, or mixtures thereof), to methanesulfonyl chloride and a base. Epoxide formation can be affected by the base-mediated (e.g., KHCO3, K2CO3, CS2CO3, or Na2CC>3, or the like) ring closure of compounds 2-7b (or 2- 7d, the enantiomer of 2-7b, or mixtures thereof) or 2-7a (or 2-7c, the enantiomer of 2-7a, or mixtures thereof) to provide epoxides 4* (or 4c*, the enantiomer of 4*, or mixtures thereof) and 5* (or 5-b*, the enantiomer of 5*, or mixtures thereof). The epoxides can then be converted into amino-alcohols 4b (or 4c, the enantiomer of 4b, or mixtures thereof) and 1-6* (or 1-7*, the enantiomer of 1-6*, or mixtures thereof) through ammonia-mediated epoxide opening using ammonia in a suitable solvent (e.g., MeOH, EtOH, or water). Subsequent treatment with TMS-azide in the presence of trimethylorthoformate and sodium acetate in acetic acid would yield compounds 6 (or 6a, the enantiomer of 6, or mixtures thereof) or 1 (or la, the enantiomer of 1, or mixtures thereof) (US 4,426,531).
Scheme 3. Synthesis of 1 via Asymmetric Dihydroxylation Method


Y is -OS02-alkyl, -OS02-substituted alkyl, -OS02-aryl, -OS02- substituted aryl, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, – 0(C=0)-aryl, -0(C=0)-substituted aryl, or halogen

R-i = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, -0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, -0(S02)-aryl, or -0(S02)-substituted aryl.
Compound 1 (or la, the enantiomer of 1, or mixtures thereof) prepared by any of the methods presented herein can be converted to a sulfonic salt of formula IX (or IXa, the enantiomer of IX, or mixtures thereof), as shown in Scheme 4. This can be accomplished by a) combining compound 1 (or la, the enantiomer of 1, or mixtures thereof), a crystallization solvent or crystallization solvent mixture (e.g., EtOAc, i‘PrOAc, EtOH, MeOH, or acetonitrile, or o
Z-S-OH
combinations thereof), and a sulfonic acid o (e.g., Z = Ph, p-tolyl, Me, or Et), b) diluting the mixture with an appropriate crystallization co-solvent or crystallization co-solvent mixture (e.g., pentane, methyl i-butylether, hexane, heptane, or toluene, or combinations thereof), and c) filtering the mixture to obtain a sulfonic acid salt of formula IX (or IXa, the enantiomer of IX, or mixtures thereof). cheme 4. Synthesis of a Sulfonic Acid Salt of Compound 1 or la

Column: Waters XBridge Shield RP18, 4.6 x 150 mm, 3.5 μιη
Mobile Phase: A = 0.05% TFA/H20, B = 0.05% TFA/ACN
Autosampler flush: 1 : 1 ACN/H20
Diluent: 1:1 ACN/H20
Flow Rate: 1.0 ml/min
Temperature: 45 °C
Detector: UV 275 nm
Pump Parameters:

EXAMPLE 1
Preparation of ethyl 2-(5-bromopyridin-2-yl)-2,2-difluoroacetate (2-Br)

2-Br Dialkylated impurity In a clean multi-neck round bottom flask, copper powder (274.7 g, 2.05 eq) was suspended in dimethyl sulfoxide (3.5 L, 7 vol) at 20 – 35 °C. Ethyl bromodifluoroacetate (449 g, 1.05 eq) was slowly added to the reaction mixture at 20 – 25 °C and stirred for 1 – 2 h. 2, 5- dibromopyridine (500 g, 1 eq) was added to the reaction mixture and the temperature was increased to 35 – 40 °C. The reaction mixture was maintained at this temperature for 18 – 24 h and the reaction progress was monitored by GC.
After the completion of the reaction, ethyl acetate (7 L, 14 vol) was added to the reaction mixture and stirring was continued for 60 – 90 min at 20 – 35 °C. The reaction mixture was filtered through a Celite bed (100 g; 0.2 times w/w Celite and 1L; 2 vol ethyl acetate). The reactor was washed with ethyl acetate (6 L, 12 vol) and the washings were filtered through a Celite bed. The Celite bed was finally washed with ethyl acetate (1 L, 2 vol) and all the filtered mother liquors were combined. The pooled ethyl acetate solution was cooled to 8 – 10 °C, washed with the buffer solution (5 L, 10 vol) below 15 °C (Note: The addition of buffer solution was exothermic in nature. Controlled addition of buffer was required to maintain the reaction mixture temperature below 15 °C). The ethyl acetate layer was washed again with the buffer solution until (7.5 L; 3 x 5 vol) the aqueous layer remained colorless. The organic layer was washed with a 1: 1 solution of 10 % w/w aqueous sodium chloride and the buffer solution (2.5 L; 5 vol). The organic layer was then transferred into a dry reactor and the ethyl acetate was distilled under reduced pressure to get crude 2-Br.
The crude 2-Br was purified by high vacuum fractional distillation and the distilled fractions having 2-Br purity greater than 93 % (with the dialkylated not more than 2 % and starting material less than 0.5 %) were pooled together to afford 2-Br.
Yield after distillation: 47.7 % with > 93 % purity by GC (pale yellow liquid). Another 10 % yield was obtained by re-distillation of impure fractions resulting in overall yield of ~ 55 – 60 %.
*H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz): 8.85 (1H, d, 1.6 Hz), 8.34 (1H, dd, J = 2.0 Hz, 6.8 Hz), 7.83 (1H, d, J = 6.8 Hz), 4.33 (2H, q, J = 6.0 Hz), 1.22 (3H, t, J = 6.0 Hz). 13C NMR: 162.22 (i, -C=0), 150.40 (Ar-C-), 149.35 (t, Ar-C), 140.52 (Ar-C), 123.01 (Ar-C), 122.07 (Ar-C), 111.80 (t, -CF2), 63.23 (-OCH2-), 13.45 (-CH2CH3).
EXAMPLE 2
Preparation of2-( 5-bromopyridin-2-yl )-l -(2,4-difluorophenyl )-2, 2-difluoroethanone ( 3-Br ) A. One-step Method
 l-Bromo-2,4-difluorobenzene (268.7 g; 1.3 eq) was dissolved in methyl tert butyl ether (MTBE, 3.78 L, 12.6 vol) at 20 – 35 °C and the reaction mixture was cooled to -70 to -65 °C using acetone/dry ice bath. n-Butyl lithium (689 rriL, 1.3 eq; 2.5 M) was then added to the reaction mixture maintaining the reaction temperature below -65 °C (Note: Controlled addition of the n-Butyl Lithium to the reaction mixture was needed to maintain the reaction mixture temperature below – 65 °C). After maintaining the reaction mixture at this temperature for 30 – 45 min, 2-Br (300 g, 1 eq) dissolved in MTBE (900 rriL, 3 vol) was added to the reaction mixture below – 65 °C. The reaction mixture was continued to stir at this temperature for 60 – 90 min and the reaction progress was monitored by GC.
l-Bromo-2,4-difluorobenzene (268.7 g; 1.3 eq) was dissolved in methyl tert butyl ether (MTBE, 3.78 L, 12.6 vol) at 20 – 35 °C and the reaction mixture was cooled to -70 to -65 °C using acetone/dry ice bath. n-Butyl lithium (689 rriL, 1.3 eq; 2.5 M) was then added to the reaction mixture maintaining the reaction temperature below -65 °C (Note: Controlled addition of the n-Butyl Lithium to the reaction mixture was needed to maintain the reaction mixture temperature below – 65 °C). After maintaining the reaction mixture at this temperature for 30 – 45 min, 2-Br (300 g, 1 eq) dissolved in MTBE (900 rriL, 3 vol) was added to the reaction mixture below – 65 °C. The reaction mixture was continued to stir at this temperature for 60 – 90 min and the reaction progress was monitored by GC.
The reaction was quenched by slow addition of 20 % w/w ammonium chloride solution (750 mL, 2.5 vol) below -65 °C. The reaction mixture was gradually warmed to 20 – 35 °C and an additional amount of 20 % w/w ammonium chloride solution (750 mL, 2.5 vol) was added. The aqueous layer was separated, the organic layer was washed with a 10 % w/w sodium bicarbonate solution (600 mL, 2 vol) followed by a 5 % sodium chloride wash (600 mL, 2 vol). The organic layer was dried over sodium sulfate (60 g; 0.2 times w/w), filtered and the sodium sulfate was washed with MTBE (300 mL, 1 vol). The organic layer along with washings was distilled below 45 °C under reduced pressure until no more solvent was collected in the receiver. The distillation temperature was increased to 55 – 60 °C, maintained under vacuum for 3 – 4 h and cooled to 20 – 35 °C to afford 275 g (73.6 % yield, 72.71 % purity by HPLC) of 3-Br as a pale yellow liquid.
*H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz):8.63 (1H, d, 1.6 Hz, Ar-H), 8.07 – 8.01 (2H, m, 2 x Ar-H), 7.72 (1H, d, J = 6.8 Hz, Ar-H), 7.07 – 6.82 (1H, m, Ar-H), 6.81 – 6.80 (1H, m, Ar-H). 13C NMR: 185.60 (t, -C=0), 166.42 (dd, Ar-C-), 162.24 (dd, Ar-C),
150.80 (Ar-C), 150.35 (Ar-C), 140.02 (Ar-C), 133.82 (Ar-C), 123.06 (Ar-C), 1122.33 (Ar-C), 118.44 (Ar-C), 114.07 (-CF2-), 122.07 (Ar-C), 105.09 (Ar-C).
B. Two-step Method via 2b-Br

2-Br (147.0 g) was dissolved in n-heptane (1.21 L) and transferred to a 5-L reactor equipped with overhead stirrer, thermocouple, condenser and addition funnel. Morpholine (202 ml) was added. The solution was heated to 60 °C and stirred overnight. The reaction was complete by HPLC analysis (0.2% 2-Br; 94.7% 2b-Br). The reaction was cooled to room temperature and 1.21 L of MTBE was added. The solution was cooled to ~4 °C and quenched by slow addition of 30% citric acid (563 ml) to maintain the internal temperature <15 °C. After stirring for one hour the layers were allowed to settle and were separated (Aq. pH=5). The organic layer was washed with 30% citric acid (322 ml) and 9% NaHC03 (322 ml, aq. pH 7+ after separation). The organic layer was concentrated on the rotary evaporator (Note 1) to 454 g (some precipitation started immediately and increased during concentration). After stirring at room temperature the suspension was filtered and the product cake was washed with n-heptane (200 ml). The solid was dried in a vacuum oven at room temperature to provide 129.2 g (77%) dense powder. The purity was 96.5% by HPLC analysis.
To a 1-L flask equipped with overhead stirring, thermocouple, condenser and addition funnel was added magnesium turnings (14.65 g), THF (580 ml) and l-bromo-2,4-difluorobenzene (30.2 g, 0.39 equiv). The mixture was stirred until the reaction initiated and self-heating brought the reaction temperature to 44 °C. The temperature was controlled with a cooling bath as the remaining l-bromo-2,4-difluorobenzene (86.1 g, 1.11 equiv) was added over about 30 min. at an internal temperature of 35-40 °C. The reaction was stirred for 2 hours while gradually cooling to room temperature. The dark yellow solution was further cooled to 12 °C.
During the Grignard formation, a jacketed 2-L flask equipped with overhead stirring, thermocouple, and addition funnel was charged with morpholine amide 2b-Br (129.0 g) and THF (645 ml). The mixture was stirred at room temperature until the solid dissolved, and then the solution was cooled to -8.7 °C. The Grignard solution was added via addition funnel over about 30 min. at a temperature of -5 to 0 °C. The reaction was stirred at 0 °C for 1 hour and endpointed by HPLC analysis. The reaction mixture was cooled to -5 °C and quenched by slow addition of 2N HC1 over 1 hour at <10 °C. The mixture was stirred for 0.5 h then the layers were allowed to settle and were separated. The aqueous layer was extracted with MTBE (280 ml). The combined organic layers were washed with 9% NaHCC>3 (263 g) and 20% NaCl (258 ml). The organic layer was concentrated on the rotary evaporator with THF rinses to transfer all the solution to the distillation flask. Additional THF (100 ml) and toluene (3 x 100 ml) were added and distilled to remove residual water from the product. After drying under vacuum, the residue was 159.8 g of a dark brown waxy solid (>theory). The purity was approximately 93% by HPLC analysis.
EXAMPLE 3
Preparation of 3-amino-l-(5-bromopyridin-2-yl)-2-(2,4-difluorophenyl)-l,l-difluoropropan- -ol (±ib-Br)

4-Br (200g, 1 eq) was added into methanolic ammonia (8.0 L; 40 vol; ammonia content: 15 – 20 % w/v) in an autoclave at 10 – 20 °C. The reaction mixture was gradually heated to 60 – 65 °C and at 3 – 4 kg/cm2 under sealed conditions for 10 – 12 h. The reaction progress was monitored by GC. After completion of the reaction, the reaction mixture was cooled to 20 – 30 °C and released the pressure gradually. The solvent was distilled under reduced pressure below 50 °C and the crude obtained was azeotroped with methanol (2 x 600 mL, 6 vol) followed by with isopropanol (600 mL, 2 vol) to afford 203 g (96.98 % yield, purity by HPLC: 94.04 %) of +4b-Br. EXAMPLE 4
Preparation of3-amino-l-(5-bromopyridin-2-yl)-2-(2,4-difluorophenyl)-l,l-difluoropropan- -ol (4b-Br or 2c-Br)

Amino alcohol ±4b-Br (150 g, 1 eq) was dissolved in an isopropanol /acetonitrile mixture (1.5L, 8:2 ratio, 10 vol) and Di-p-toluoyl-L-tartaric acid (L-DPTTA) (84.05 g, 0.55 eq) was added into the reactor at 20 – 30 °C. The reaction mixture was heated to 45 – 50 °C for 1 – 1.5 h (Note: The reaction mixture becomes clear and then became heterogeneous). The reaction mixture was gradually cooled to 20 – 30 °C and stirred for 16 – 18 h. The progress of the resolution was monitored by chiral HPLC analysis.
After the completion of the resolution, the reaction mixture was gradually cooled to 20 – 35 °C. The reaction mixture was filtered and the filtered solid was washed with a mixture of acetonitrile and isopropanol (8:2 mixture, 300 mL, 2 vol) and dried to afford 75 g of the L- DPTTA salt (95.37 % ee). The L-DPTTA salt obtained was chirally enriched by suspending the salt in isopropanol /acetonitrile (8:2 mixture; 750 mL, 5 vol) at 45 – 50 °C for 24 – 48 h. The chiral enhancement was monitored by chiral HPLC; the solution was gradually cooled to 20 – 25 °C, filtered and washed with an isoporpanol /acetonitrile mixture (8:2 mixture; 1 vol). The purification process was repeated and after filtration, the salt resulted in chiral purity greater than 96 % ee. The filtered compound was dried under reduced pressure at 35 – 40 °C to afford 62 g of the enantio-enriched L-DPPTA salt with 97.12% ee as an off-white solid. The enantio-enriched L-DPTTA salt (50 g, 1 eq) was dissolved in methanol (150 mL, 3 vol) at 20 – 30 °C and a potassium carbonate solution (18.05 g K2CO3 in 150 mL water) was slowly added at 20 – 30 °C under stirring. The reaction mixture was maintained at this temperature for 2 – 3 h (pH of the solution at was maintained at 9). Water (600 mL, 12 vol) was added into the reaction mixture through an additional funnel and the reaction mixture was stirred for 2 – 3 h at 20 – 30 °C. The solids were filtered; washed with water (150 mL, 3 vol) and dried under vacuum at 40 – 45 °C to afford 26.5 g of amino alcohol 4b-Br or 4c-Br with 99.54 % chemical purity, 99.28 % ee as an off-white solid. (Water content of the chiral amino alcohol is below 0.10 % w/w).
1H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz):8.68 (1H, d, J = 2.0 Hz, Ar- H), 8.16 (1H, dd, J = 8.0 Hz, 2.0 Hz, Ar-H), 7.49 – 7.43 (1H, m, Ar-H), 7.40 (1H, d, J = 8 Hz, Ar-H), 7.16 – 7.11 (1H, m, Ar-H), 7.11 – 6.99 (1H, m, Ar-H), 3.39 – 3.36 (1H, m, -OCHAHB– ), 3.25 – 3.22 (1H, m, -OCHAHB-).13C NMR: 163.87 -158.52 (dd, 2 x Ar-C-), 150.88 (Ar-C), 149.16 (Ar-C), 139.21 (Ar-C), 132.39 (Ar-C), 124.49 (Ar-C), 122.17 (Ar-C), 121.87 (d, Ar- C), 119.91 (t, -CF2-), 110.68 (Ar-C), 103.97 (i, Ar-C), 77.41 (i,-C-OH), 44.17 (-CH2-NH2).
EXAMPLE 5
Preparation of l-(5-bromopyridin-2-yl)-2-(2,4-difluorophenyl)-l,l-difluoro-3-(lH-tetrazol-l- yl)propan-2-ol (l-6*-Br or l-7*-Br)

4b-Br or 4c-Br (20.0 g, 1 eq.) was added to acetic acid (50 mL, 2.5 vol) at 25 – 35 °C followed by the addition of anhydrous sodium acetate (4.32 g, 1 eq), trimethyl orthoformate (15.08 g, 2.7 eq). The reaction mixture was stirred for 15 – 20 min at this temperature and trimethylsilyl azide (12.74 g, 2.1 eq) was added to the reaction mixture (Chilled water was circulated through the condenser to minimize the loss of trimethylsilyl azide from the reaction mixture by evaporation). The reaction mixture was then heated to 70 – 75 °C and maintained at this temperature for 2 -3 h. The reaction progress was monitored by HPLC. Once the reaction was complete, the reaction mixture was cooled to 25 – 35 °C and water (200 mL, 10 vol) was added. The reaction mixture was extracted with ethyl acetate (400 mL, 20 vol) and the aqueous layer was back extracted with ethyl acetate (100 mL, 5 vol). The combined organic layers were washed with 10 % potassium carbonate solution (3 x 200 mL; 3 x 10 vol) followed by a 10 % NaCl wash (1 x 200 mL, 10 vol). The organic layer was distilled under reduced pressure below 45 °C. The crude obtained was azeotroped with heptanes (3 x 200 mL) to get 21.5g (94 % yield, 99.26 5 purity) of tetrazole 1-6* or 1-7* compound as pale brown solid (low melting solid).
1H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz NMR instrument): 9.13 (1H, Ar-H), 8.74 (1H, Ar-H), 8.22 – 8.20 (1H, m, Ar-H), 7.44 (1H, d, J = 7.2 Hz, Ar-H), 7.29 (1H„
Ar-H), 7.23 – 7.17 (1H, m, Ar-H), 6.92 – 6.88 (1H, Ar-H), 5.61 (1H, d, J = 1 1.2 Hz, – OCHAHB-), 5.08 (1H, d, J = 5.6 Hz, -OCHAHB-).13C NMR: 163.67 -161.59 (dd, Ar-C-), 160.60 – 158.50 (dd, Ar-C-), 149.65 (Ar-C), 144.99 (Ar-C), 139.75 (Ar-C), 131.65 (Ar-C), 124.26 (Ar-C), 122.32 (d, Ar-C), 119.16 (t, -CF2-), 118.70 (d, Ar-C), 1 11.05 (d, Ar-C) 104.29 (t, Ar-C), 76.79 (i,-C-OH), 59.72 (Ar-C), 50.23 (-OCH2N-). EXAMPLE 6
Preparation of 2-(2,4-difluorophenyl)-l , 1 -difluoro-3-( 1 H-tetrazol-1 -yl)-l -(5-(4-(2,2,2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (1 or la)
A. Preparation of 1 or la via l-6*-Br or l-7*-Br

Synthesis of 4,4,5, 5-tetramethyl-2-(4-(2,2,2-trifluoroethoxy)phenyl)-l,3,2-dioxaborolane Potassium carbonate (59.7 g, 2.2 eq.) was added to a slurry of DMF (190 mL, 3.8 Vol.), 4- Bromo phenol (37.4g, 1.1 eq.) and 2,2,2-trifluroethyl tosylate (50.0 g, 1.0 eq.) at 20 – 35 °C under an inert atmosphere. The reaction mixture was heated to 115 – 120 °C and maintained at this temperature for 15 – 18 h. The reaction progress was monitored by GC. The reaction mixture was then cooled to 20 – 35 °C, toluene (200 mL, 4.0 vol.) and water (365 mL, 7. 3 vol.) were added at the same temperature, stirred for 10 – 15 minutes and separated the layers. The aqueous layer was extracted with toluene (200 mL, 4.0 vol.). The organic layers were combined and washed with a 2M sodium hydroxide solution (175 mL, 3.5 vol.) followed by a 20 % sodium chloride solution (175 mL, 3.5 vol.). The organic layer was then dried over anhydrous sodium sulfate and filtered. The toluene layer was transferred into clean reactor, spurged with argon gas for not less than 1 h. Bis(Pinacolato) diborane (47 g, 1.1 eq.), potassium acetate (49.6 g, 3.0 eq.) and 1,4-dioxane (430 mL, 10 vol.) were added at 20 -35 °C, and spurged the reaction mixture with argon gas for at least 1 h. Pd(dppf)Cl2 (6.88 g, 0.05eq) was added to the reaction mixture and continued the argon spurging for 10 – 15 minutes. The reaction mixture temperature was increased to 70 – 75 °C, maintained the temperature under argon atmosphere for 15 – 35 h and monitored the reaction progress by GC. The reaction mixture was cooled to 20 – 35 °C, filtered the reaction mixture through a Celite pad, and washed with ethyl acetate (86 mL, 2 vol.). The filtrate was washed with water (430 mL, 10 vol.). The aqueous layer was extracted with ethyl acetate (258 mL, 6 vol.) and washed the combined organic layers with a 10 % sodium chloride solution (215 mL, 5 vol.). The organic layer was dried over anhydrous sodium sulfate (43g, 1 time w/w), filtered and concentrated under reduced pressure below 45 °C to afford crude 4,4,5, 5-tetramethyl-2-(4-(2,2,2- trifluoroethoxy)phenyl)-l,3,2-dioxaborolane (65 g; 71 % yield with the purity of 85.18 % by GC). The crude 4,4,5,5-tetramethyl-2-(4-(2,2,2-trifluoroethoxy)phenyl)-l,3,2-dioxaborolane (65 g, 1 eq.) was dissolved in 10 % ethyl acetate – n-Heptane (455 mL, 7 vol.) and stirred for 30 – 50 minutes at 20 – 35 °C. The solution was filtered through a Celite bed and washed with 10 % ethyl acetate in n-Heptane (195 mL, 3 vol.). The filtrate and washings were pooled together, concentrated under vacuum below 45 °C to afford 4,4,5, 5-tetramethyl-2-(4-(2,2,2- trifluoroethoxy)phenyl)-l,3,2-dioxaborolane as a thick syrup (45.5 g; 70 % recovery). This was then dissolved in 3 % ethyl acetate-n-heptane (4 vol.) and adsorbed on 100 – 200 M silica gel (2 times), eluted through silica (4 times) using 3 % ethyl acetate – n- heptane. The product rich fractions were pooled together and concentrated under vacuum. The column purified fractions (> 85 % pure) were transferred into a round bottom flask equipped with a distillation set-up. The compound was distilled under high vacuum below 180 °C and collected into multiple fractions. The purity of fractions was analyzed by GC (should be > 98 % with single max impurity < 1.0 %). The less pure fractions (> 85 % and < 98 % pure fraction) were pooled together and the distillation was repeated to get 19g (32% yield) of 4,4,5, 5-tetramethyl-2-(4- (2,2,2-trifluoroethoxy)phenyl)-l,3,2-dioxaborolane as a pale yellow liquid.
*H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz):7.64 (2H, d, 6.8 Hz), 7.06 (2H, d, J = 6.4 Hz), 4.79 (2H, q, J = 6.8 Hz), 1.28 (12H, s).
13C NMR: 159.46 (Ar-C-O-), 136.24 (2 x Ar-C-), 127.77 – 120.9 (q, -CF3), 122.0 (Ar-C-B), 114.22 (2 x Ar-C-), 64.75 (q, J = 27.5 Hz).
Synthesis of 2-(2.4-difluorophenyl)-l.l-difluoro-3-(lH-tetrazol-l-yl)-l-(5-(4-(2.2.2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (1 or la)
l-6*-Br or l-7*-Br (14 g, 0.03 mol, 1 eq) was added to tetrahydrofuran (168 mL, 12 vol) at 25 – 35 °C and the resulting solution was heated to 40 – 45 °C. The reaction mixture was maintained at this temperature for 20 – 30 min under argon bubbling. Sodium carbonate (8.59 g, 0.08 mol, 2.5 eq) and water (21 mL, 1.5 vol) were added into the reaction mixture and the bubbling of argon was continued for another 20 – 30 min. 4,4,5, 5-tetramethyl-2-(4-(2,2,2- trifluoroethoxy)phenyl)-l,3,2-dioxaborolane (10.76 g, 1.1 eq) dissolved in tetrahydrofuran (42 mL, 3 vol) was added into the reaction mixture and argon bubbling was continued for 20 – 30 min. Pd(dppf)Cl2 (2.65 g, 0.1 eq) was added to the reaction mixture under argon bubbling and stirred for 20 – 30 min (Reaction mixture turned into dark red color). The reaction mixture was heated to 65 – 70 °C and maintained at this temperature for 3 – 4 h. The reaction progress was monitored by HPLC. The reaction mixture was cooled to 40 – 45 °C and the solvent was distilled under reduced pressure. Toluene (350 mL, 25 vol.) was added to the reaction mixture and stirred for 10 – 15 min followed by the addition of water (140 mL, 10 vol). The reaction mixture was filtered through Hyflo (42 g, 3 times), the layers were separated and the organic layer was washed with water (70 mL, 5 vol) and a 20 % w/w sodium chloride solution (140 mL, 10 vol). The organic layer was treated with charcoal (5.6 g, 0.4 times, neutral chalrcoal), filtered through Hyflo. (lS)-lO-Camphor sulfonic acid (7.2 g, 1 eq.) was added to the toluene layer and the resulting mixture was heated to 70 – 75 °C for 2 – 3 h. The reaction mixture was gradually cooled to 25 – 35 °C and stirred for 1 – 2 h. The solids were filtered, washed with toluene (2 x 5 vol.) and then dried under vacuum below 45 °C to afford 18.0 g of an off white solid. The solids (13.5 g, 1 eq.) were suspended in toluene (135 mL, 10 vol) and neutralized by adding 1M NaOH solution (1.48 vol, 1.1 eq) at 25 – 35 °C and stirred for 20 – 30 min. Water (67.5 mL, 5 vol) was added to the reaction mixture and stirred for 10 – 15 min, and then the layers were separated. The organic layer was washed with water (67.5 mL, 5 vol) to remove the traces of CSA. The toluene was removed under reduced pressure below 45 °C to afford crude 1 or la. Traces of toluene were removed by azeotroping with ethanol (3 x 10 vol), after which light brown solid of crude 1 or la (7.5 g, 80% yield) was obtained.
The crude 1 or la (5 g) was dissolved in ethanol (90 mL, 18 vol.) at 20 – 35 °C, and heated to 40 – 45 °C. Water (14 vol) was added to the solution at 40 – 45 °C, the solution was maintained at this temperature for 30 – 45 min and then gradually cooled to 20 – 35 °C. The resulting suspension was continued to stir for 16 – 18 h at 20 – 35 °C, an additional amount of water (4 vol.) was added and the stirring continued for 3 – 4 h. The solids were filtered to afford 4.0 g (80% recovery) of 1 or la (HPLC purity >98%) as an off-white solid.
1H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz):9.15 (1H, s, Ar-H), 8.93 (1H, d, J = 0.8 Hz, Ar-H), .8.22 – 8.20 (1H, m, Ar-H), 7.80 (2H, d, J = 6.8 Hz, Ar-H), 7.52 (1H, d, J = 6.8 Hz, Ar-H), 7.29 (1H, d,J = 3.2Hz, Ar-H), 7.27 – 7.21 (1H, m, Ar-H), 7.23 – 7.21 (2H, d, J = 6.8 Hz, Ar-H), 7.19 (1H, d, J = 6.8 Hz, Ar-H), 6.93 – 6.89 (1H, m, Ar-H), 5.68 (1H, / = 12 Hz, -CHAHB), 5.12 (2H, d, J = 11.6 Hz, -CHAHB), 4.85 (2H, q, J = 1.6 Hz).
13C NMR: 163.93 – 158.33 (m, 2 x Ar-C), 157.56 (Ar-C), 149.32 (i, Ar-C), 146.40 (Ar-C), 145.02 (Ar-C), 136.20 (Ar-C), 134.26 (2 x Ar-C), 131.88 – 131.74 (m, AR-C), 129.72 (Ar-C), 128.47 (2 x Ar-C), 123.97 (q, -CF2-), 122.41 (Ar-C), 119.30 (-CF3), 118.99 (Ar-C), 115.65 (2 x Ar-C), 110.99 (d, Ar-C), 104.22 (i, Ar-C), 77.41 – 76.80 (m, Ar-C), 64.72 (q, -OCH2-CF3), 50.54 (-CH2-N-).
B. Preparation of 1 or la via 4b-Br or 4c-Br


Synthesis of 3-amino-2-(2.4-difluorophenyl)-l.l-difluoro-l-(5-(4-(2.2.2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (8a or 8b)
Potassium carbonate (30.4 g) and water (53.3 g) were charged to a 1-L flask equipped with overhead stirring, thermocouple, and nitrogen/vacuum inlet valve, and stirred until dissolved. The boronic acid (19.37 g), a solution of 4b-Br or 4c-Br in 2-butanol (103.5 g, 27.8 g theoretical 4b-Br or 4c-Br)) and 2-BuOH (147.1 g) were added and stirred to form a clear mixture. The flask was evacuated and refilled with nitrogen 3 times. Pd(d f)2Cl2 (0.30 g) was added and stirred to form a light orange solution. The flask was evacuated and refilled with nitrogen 4 times. The mixture was heated to 85 °C and stirred overnight and endpointed by HPLC analysis. The reaction mixture was cooled to 60 °C and the layers were allowed to settle. The aqueous layer was separated. The organic layer was washed with 5% NaCl solution (5 x 100 ml) at 30-40 °C. The organic layer was filtered and transferred to a clean flask with rinses of 2-BuOH. The combined solution was 309.7 g, water content 13.6 wt% by KF analysis. The solution was diluted with 2-BuOH (189 g) and water (10 g). Theoretically the solution contained 34.8 g product, 522 ml (15 volumes) of 2-BuOH, and 52.2 ml (1.5 volumes) of water. L-Tartaric acid (13.25 g) was added and the mixture was heated to a target temperature of 70-75 °C. During the heat-up, a thick suspension formed. After about 15 minutes at 70-72 °C the suspension became fluid and easily stirred. The suspension was cooled at a rate of 10 °C/hour to 25 °C then stirred at 25 °C for about 10 hours. The product was collected on a vacuum filter and washed with 10:1 (v/v) 2-BuOH/water (50 ml) and 2- butanol (40 ml). The salt was dried in a vacuum oven at 60 °C with a nitrogen purge for 2 days. The yield was 40.08 g of 8a or 8b as a fluffy, grayish-white solid. The water content was 0.13 wt% by KF analysis. The yield was 87.3% with an HPLC purity of 99.48%. Synthesis of 2-(2,4-difluorophenyl)-l,l-difluoro-3-(lH-tetrazol-l-yl)-l-(5-(4-(2,2,2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (1 or la)
To a 350 ml pressure bottle were charged acetic acid (73 ml), 8a or 8b (34.8 g), sodium acetate (4.58 g) and trimethylorthoformate (16.0 g). The mixture was stirred for 18 min. at room temperature until a uniform suspension was obtained. Azidotrimethylsilane (8.88 g) was added and the bottle was sealed. The bottle was immersed in an oil bath and magnetically stirred. The oil bath was at 52 °C initially, and was warmed to 62-64 °C over about ½ hour. The suspension was stirred at 62-64 °C overnight. After 20.5 hours the suspension was cooled to room temperature and sampled. The reaction was complete by HPLC analysis. The reaction was combined with three other reactions that used the same raw material lots and general procedure (total of 3.0 g additional starting material). The combined reactions were diluted with ethyl acetate (370 ml) and water (368 ml) and stirred for about ½ hour at room temperature. The layers were settled and separated. The organic layer was washed with 10% K2C03 solution (370 ml/ 397 g) and 20% NaCl solution (370 ml/ 424 g). The organic layer (319 g) was concentrated, diluted with ethanol (202 g) and filtered, rinsed with ethanol (83 g). The combined filtrate was concentrated to 74 g of amber solution.
The crude 1 or la solution in ethanol (74 g solution, containing theoretically 31.9 g 1 or la) was transferred to a 2-L flask equipped with overhead stirring, thermocouple, and addition funnel. Ethanol (335 g) was added including that used to complete the transfer of the 1 or la solution. The solution was heated to nominally 50 °C and water (392 g) was added over 12 minutes. The resulting hazy solution was seeded with 1 or la crystals and stirred at 50 °C. After about ½ hour the mixture was allowed to cool to 40 °C over about ½ hour during which time crystallization started. Some darker colored chunky solid separated out from the main suspension. The pH of the crystallizing mixture was adjusted from 4.5 to 6 using 41% KOH (1.7 g). After about 1 hour a good suspension had formed. Additional water (191 g) was added slowly over ½ hour. The suspension was heated to 50 °C and cooled at 5 °C/min to room temperature. After stirring overnight the suspension was cooled in a water bath to 16 °C and filtered after 1 hour. The wet cake was washed with 55:45 (v/v) water/ethanol (2 x 50 ml) and air-dried on the vacuum filter funnel overnight. Further drying at 40 °C in a vacuum oven with a nitrogen bleed resulted in no additional weight loss. The yield was 30.2 g of off-white fine powder plus some darker granular material. By in-process HPLC analysis there was no difference in the chemical purity of the darker and lighter materials. The purity was 99.4%. The water content was 2.16 wt% by KF analysis. The residual ethanol was 1.7 wt% estimated by ‘Ft NMR analysis. The corrected yield was 29.0 g, 91.0% overall yield for tetrazole formation and crystallization. The melting point was 65 °C by DSC analysis.
FDA Approves Mycovia Pharmaceuticals’ VIVJOA™ (oteseconazole), the First and Only FDA-Approved Medication for Recurrent Vulvovaginal Candidiasis (Chronic Yeast Infection)
– Approval of VIVJOA™ marks a significant therapeutic advancement for reducing the incidence of RVVC, a condition with substantial unmet need, in permanently infertile and postmenopausal women
– VIVJOA™ is the first FDA approval in Mycovia’s pipeline of novel treatments for fungal infections
– U.S. commercial launch of VIVJOA™ expected in Q2
DURHAM, N.C.–(BUSINESS WIRE)–The U.S. Food and Drug Administration (FDA) approved VIVJOA™ (oteseconazole capsules), an azole antifungal indicated to reduce the incidence of recurrent vulvovaginal candidiasis (RVVC) in females with a history of RVVC who are NOT of reproductive potential. VIVJOA is the first and only FDA-approved medication for this condition and provides sustained efficacy demonstrated by significant long-term reduction of RVVC recurrence through 50 weeks versus comparators. VIVJOA is the first FDA-approved product for Mycovia Pharmaceuticals, Inc. (Mycovia), an emerging biopharmaceutical company dedicated to recognizing and empowering those living with unmet medical needs by developing novel therapies.
RVVC, also known as chronic yeast infection, is defined by the Centers for Disease Control and Prevention (CDC) as three or more symptomatic acute episodes of yeast infection in 12 months. RVVC is a distinct condition from vulvovaginal candidiasis (VVC), and until now, there have been no FDA-approved medications specifically indicated for it. Nearly 75% of all adult women will have at least one yeast infection in their lifetime, with approximately half experiencing a recurrence. Of those women, up to 9% develop RVVC. “After nearly two decades of living with chronic yeast infection and feeling like there was no hope from the itchiness, irritation and constant dread of when the next yeast infection would return, I was overjoyed to even be a part of this clinical trial,” said Leslie Ivey, RVVC patient and clinical trial participant. “It is gratifying to see RVVC finally get the attention it deserves.” Symptoms of RVVC include vaginal itching, burning, irritation and inflammation. Some women may experience abnormal vaginal discharge and painful sexual intercourse or urination, causing variable but often severe discomfort and pain. VIVJOA’s FDA approval is based upon the positive results from three Phase 3 trials of oteseconazole – two global, pivotal VIOLET studies and one U.S.-focused ultraVIOLET study, including 875 patients at 232 sites across 11 countries. In the two global VIOLET studies, 93.3% and 96.1% of women with RVVC who received VIVJOA did not have a recurrence for the 48-week maintenance period compared to 57.2% and 60.6% of patients who received placebo (p <0.001). In the ultraVIOLET study, 89.7% of women with RVVC who received VIVJOA cleared their initial yeast infection and did not have a recurrence for the 50-week maintenance period compared to 57.1% of those who received fluconazole followed by placebo (p <0.001). The most common side effects reported in Phase 3 clinical studies were headache (7.4%) and nausea (3.6%). VIVJOA is contraindicated in those with a hypersensitivity to oteseconazole, and based on data from rat studies, also in females who are of reproductive potential, pregnant, or lactating. Please see additional Important Safety Information below. Patrick Jordan, CEO of Mycovia Pharmaceuticals and Partner at NovaQuest Capital Management, stated, “We celebrate this important milestone for Mycovia, as VIVJOA is the first antifungal in our pipeline to obtain FDA approval and achieves our goal to fulfill a previously unmet medical need among women suffering from RVVC. We are honored to lead this advancement in women’s health.” “We believe the market need for VIVJOA is strong, and we are eager to execute our commercial plans,” Jordan continued. “As we enter a new chapter of our history as a commercial biopharmaceutical company, we will continue driving our mission forward to develop novel therapies for overlooked conditions.” Oteseconazole is designed to inhibit fungal CYP51, which is required for fungal cell wall integrity, and this selective interaction is also toxic to fungi, resulting in the inhibition of fungal growth. Due to its chemical structure, oteseconazole has a lower affinity for human CYP enzymes as compared to fungal CYP enzymes. The FDA granted oteseconazole Qualified Infectious Disease Product and Fast Track designations. “A medicine with VIVJOA’s sustained efficacy combined with the clinical safety profile has been long needed, as until now, physicians and their patients have had no FDA-approved medications for RVVC,” stated Stephen Brand, Ph.D., Chief Development Officer of Mycovia. “We are excited to be the first to offer a medication designed specifically for RVVC, a challenging and chronic condition that is expected to increase in prevalence over the next decade.” Mycovia is planning its commercial launch of VIVJOA™ in the second quarter of 2022. About Recurrent Vulvovaginal Candidiasis RVVC is a debilitating, chronic infectious condition that affects 138 million women worldwide each year. RVVC, also known as chronic yeast infection, is a distinct condition from vulvovaginal candidiasis (VVC) and defined as three or more symptomatic acute episodes of yeast infection in 12 months. Primary symptoms include vaginal itching, burning, irritation and inflammation. Some women may experience abnormal vaginal discharge and painful sexual intercourse or urination, causing variable but often severe discomfort and pain. About VIVJOA™ VIVJOA™ (oteseconazole) is an azole antifungal indicated to reduce the incidence of recurrent vulvovaginal candidiasis (RVVC) in females with a history of RVVC who are NOT of reproductive potential. VIVJOA is the first and only FDA-approved medication that provides sustained efficacy demonstrated by significant long-term reduction of RVVC recurrence through 50 weeks versus comparators. Oteseconazole is designed to inhibit fungal CYP51, which is required for fungal cell wall integrity, and this selective interaction is also toxic to fungi, resulting in the inhibition of fungal growth. Due to its chemical structure, oteseconazole has a lower affinity for human CYP enzymes as compared to fungal CYP enzymes. The FDA approved VIVJOA based upon the positive results from three Phase 3 clinical trials of oteseconazole – two global, pivotal VIOLET studies and one U.S.-focused ultraVIOLET study, including 875 patients at 232 sites across 11 countries.“We believe the market need for VIVJOA is strong, and we are eager to execute our commercial plans”
Tweet this
References
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215888s000lbl.pdf
- ^ “Vivjoa: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 27 April 2022.
- ^ Jump up to:a b “FDA Approves Mycovia Pharmaceuticals’ VIVJOA (oteseconazole), the First and Only FDA-Approved Medication for Recurrent Vulvovaginal Candidiasis (Chronic Yeast Infection)” (Press release). Mycovia Pharmaceuticals. 28 April 2022. Retrieved 28 April 2022 – via Business Wire.
- ^ World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 76”. WHO Drug Information. 30 (3). hdl:10665/331020.
Further reading
- Sobel JD, Nyirjesy P (December 2021). “Oteseconazole: an advance in treatment of recurrent vulvovaginal candidiasis”. Future Microbiology. 16: 1453–1461. doi:10.2217/fmb-2021-0173. PMID 34783586.
External links
- “Oteseconazole”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03562156 for “A Study of Oral Oteseconazole for the Treatment of Patients With Recurrent Vaginal Candidiasis (Yeast Infection) (VIOLET)” at ClinicalTrials.gov
- Clinical trial number NCT03561701 for “A Study of Oral Oteseconazole (VT-1161) for the Treatment of Patients With Recurrent Vaginal Candidiasis (Yeast Infection) (VIOLET)” at ClinicalTrials.gov
- Clinical trial number NCT03840616 for “Study of Oral Oteseconazole (VT-1161) for Acute Yeast Infections in Patients With Recurrent Yeast Infections (ultraVIOLET)” at ClinicalTrials.gov
 |
|
| Clinical data | |
|---|---|
| Trade names | Vivjoa |
| Other names | VT-1161 |
| License data | |
| Routes of administration | By mouth |
| Drug class | Antifungal |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.277.989 |
| Chemical and physical data | |
| Formula | C23H16F7N5O2 |
| Molar mass | 527.403 g·mol−1 |
| 3D model (JSmol) | |


It's only fair to share...