














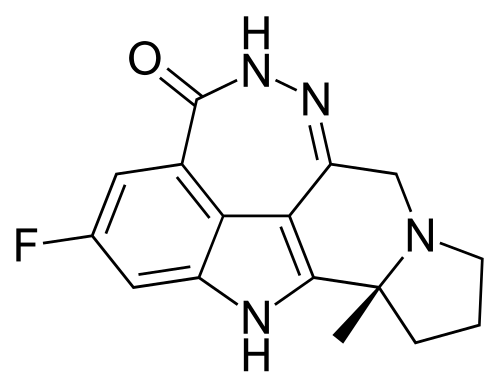
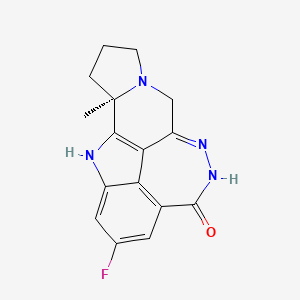
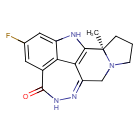
Pamiparib
BGB-290 APPROVED CHINA 2022, BEIGENE
(2R)-14-fluoro-2-methyl-6,9,10,19-tetrazapentacyclo[14.2.1.02,6.08,18.012,17]nonadeca-1(18),8,12(17),13,15-pentaen-11-one
- 1446261-44-4
- 8375F9S90C
- 5,6,7a,11-Tetraazacyclohepta(def)cyclopenta(a)fluoren-4(7H)-one, 2-fluoro-5,8,9,10,10a,11-hexahydro-10a-methyl-, (10aR)-
- 5,6,7a,11-Tetraazacyclohepta[def]cyclopenta[a]fluoren-4(7H)-one, 2-fluoro-5,8,9,10,10a,11-hexahydro-10a-methyl-, (10aR)-
- 298.31 g/mol, C16H15FN4O
Pamiparib, sold under the brand name Partruvix, is a pharmaceutical drug used for the treatment of various types of cancer. Pamiparib is a member of the PARP inhibitor drug class.[1]
In China, it is approved for the treatment of germline BRCA mutation-associated recurrent advanced ovarian, fallopian tube, and primary peritoneal cancers previously treated with two or more lines of chemotherapy.[2]
It is currently under investigation for the treatment of other forms of cancer.[3][1]
Pamiparib is under investigation in clinical trial NCT03933761 (Pamiparib in Fusion Positive, Reversion Negative High Grade Serous Ovarian Cancer or Carcinosarcoma With BRCA1/2 Gene Mutations If Progression on Substrate Poly ADP Ribose Polymerase Inhibitbor (PARPI) or Chemotherapy).
Pamiparib is an orally bioavailable inhibitor of the nuclear enzyme poly(ADP-ribose) polymerase (PARP), with potential antineoplastic activity. Upon administration, pamiparib selectively binds to PARP and prevents PARP-mediated repair of single-strand DNA breaks via the base-excision repair (BER) pathway. This enhances the accumulation of DNA strand breaks, promotes genomic instability, and eventually leads to apoptosis. PARP is activated by single-strand DNA breaks and, subsequently, catalyzes post-translational ADP-ribosylation of nuclear proteins which then transduce signals to recruit other proteins to repair damaged DNA. Pamiparib may both potentiate the cytotoxicity of DNA-damaging agents and reverse tumor cell chemo- and radioresistance.
REF
https://www.thieme-connect.com/products/ejournals/abstract/10.1055/s-0040-1719372
REF
J. Med. Chem. 2020, 63, 15541−15563.
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.0c01346
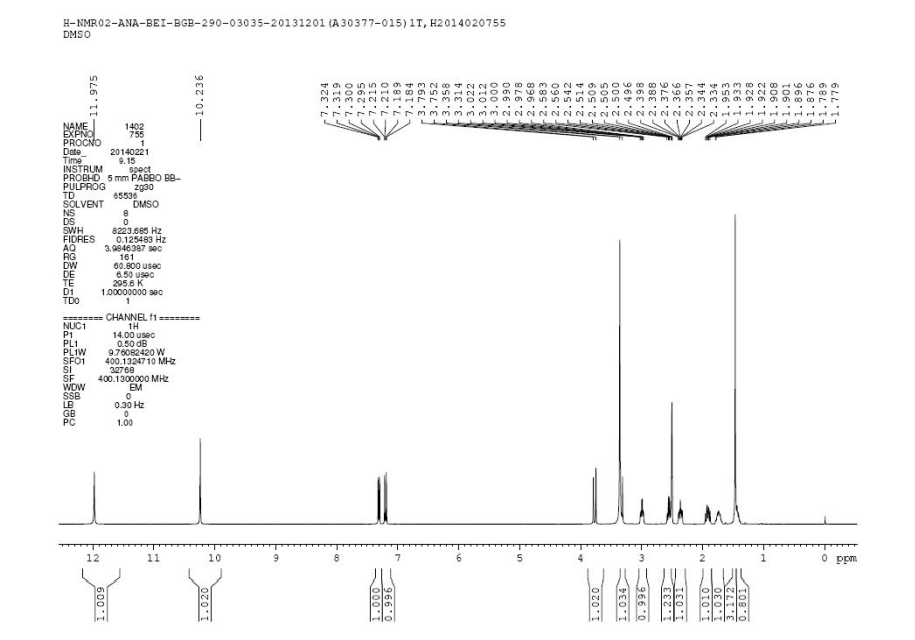
1HNMR 400, DMSO D6
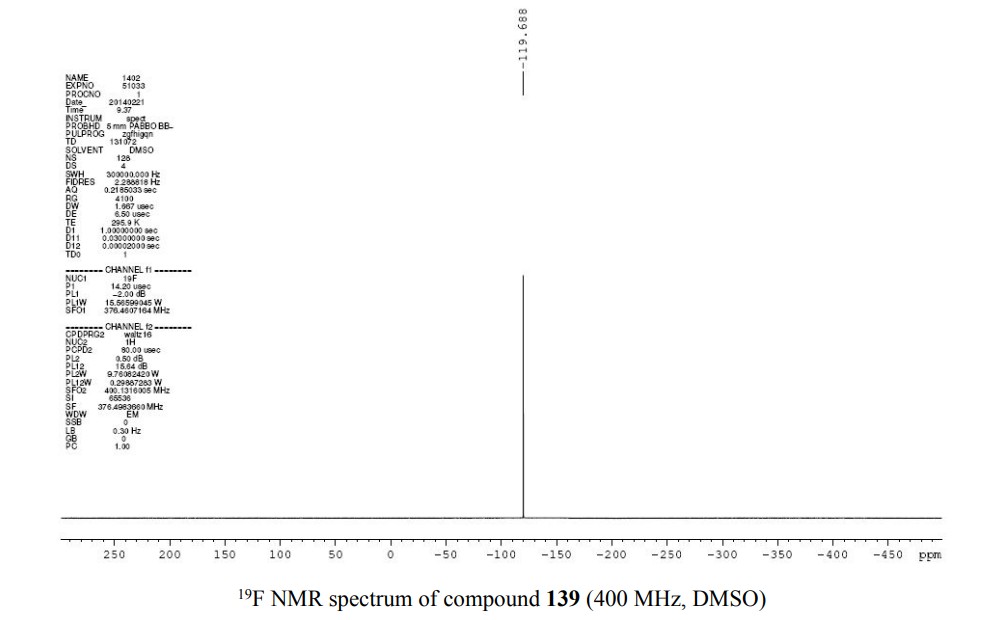
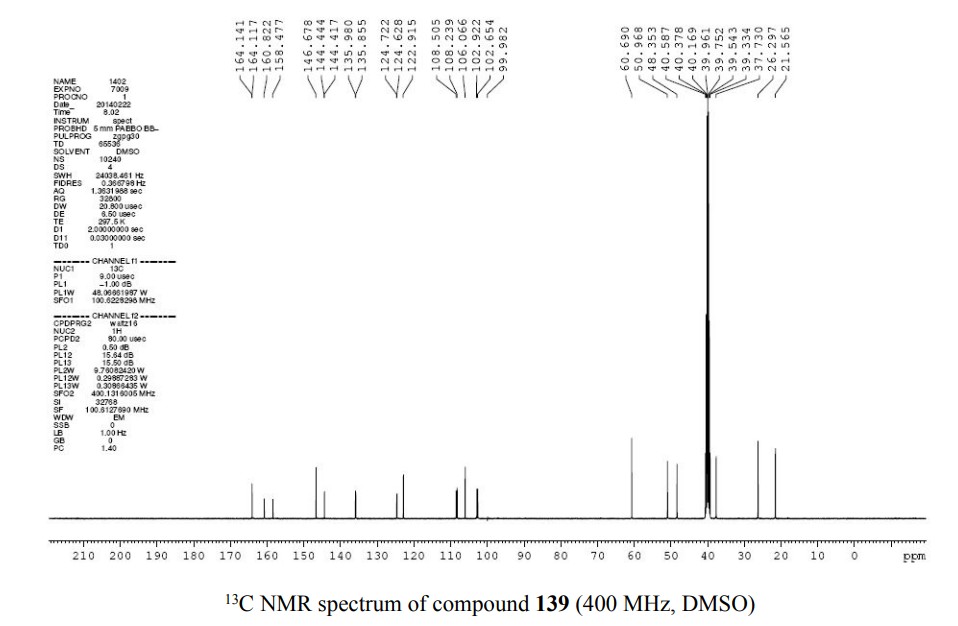
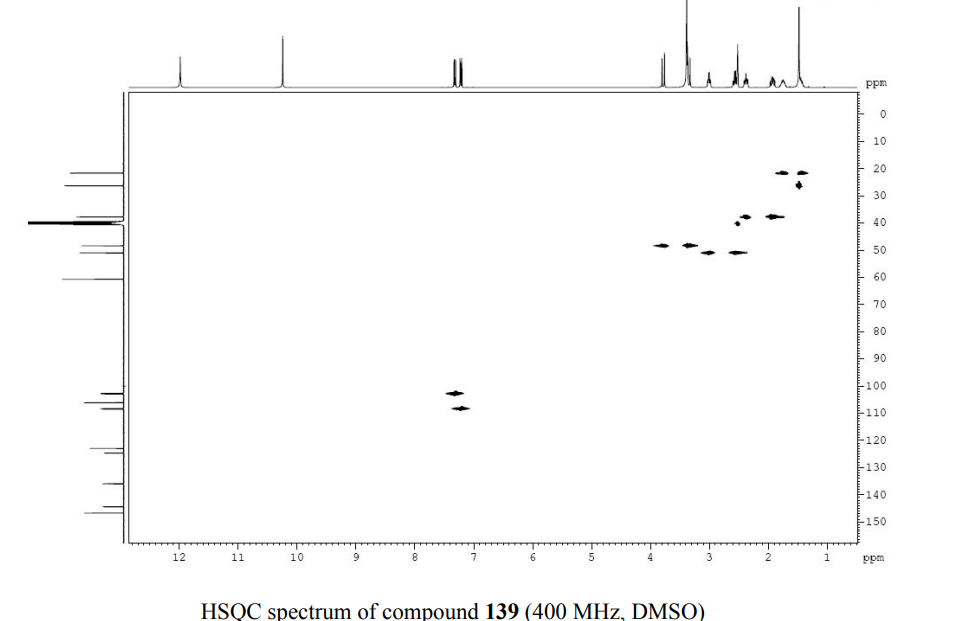
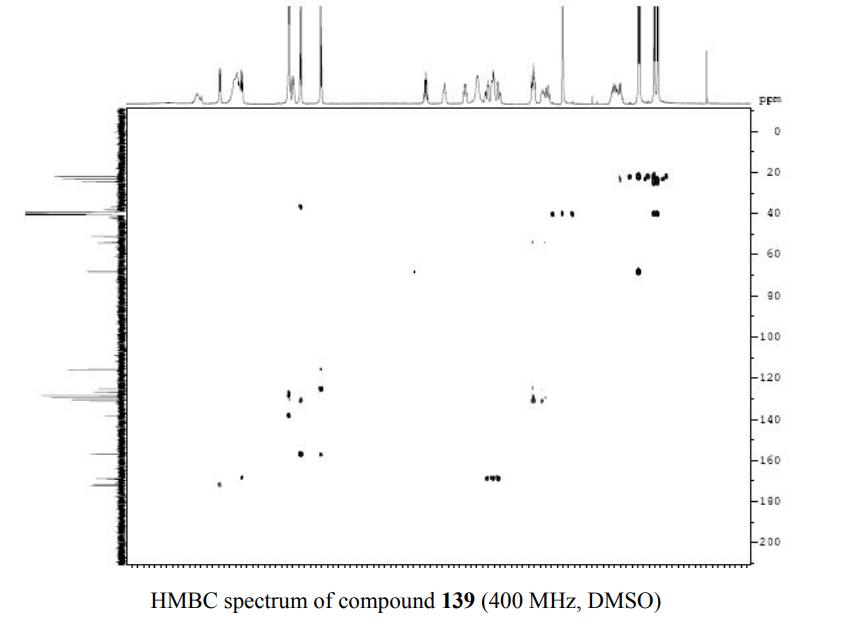
PATENT
WO 2018157794
https://patents.google.com/patent/WO2018157794A1/en
(R) -2-fluoro-10a-methyl-7, 8, 9, 10, 10a, 11-hexahydro-5, 6, 7a, 11-tetraazacyclohepta [def] cyclopenta – [a] fluoren-4 (5H) -one (hereafter Compound 1) , has been disclosed as a highly selective and potent Parp1/2 inhibitor, See WO 2013/097225 A1, which is incorporated herein by reference.

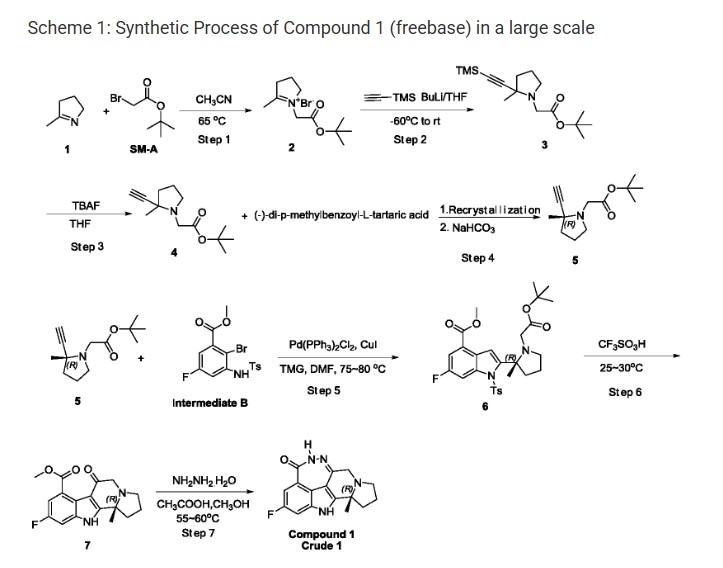
Step 1: Synthesis of Compound-2

t-Butyl bromoacetate (51.7 Kg) was dissolved in anhydrous acetonitrile (72 Kg) . The temperature was raised to 65-75 ℃, then methyl pyrroline (22 Kg) was added. The reaction mixture was condensed after the reaction was completed, the residual acetonitrile was removed by adding THF and then condensing. After GC showed a complete removal of acetonitrile, more THF was added and stirred. The resulting solid was filtered and collected. 44.1 Kg of off white solid Compound-2 was obtained. 1H NMR (400 MHz, DMSO-d6) δ 4.91 (s, 2H) , 4.15 (m, 2H) , 3.29 (m, 2H) , 2.46 (s, 3H) , ) , 2.14 (m, 2H) , 1.46 (s, 9H) ppm.
Step 2: Synthesis of Compound-3

To a cool (-60 ℃) solution of trimethylsilyl acetyne (12.4 Kg) in THF was added a solution of n-butyl lithium in hexane (43.4 Kg) . After complete addition of n-butyl lithium solution, the resulting mixture was stirred for additional 1-2 h and then the entire solution was transferred into a suspension of Compound-2 (31 Kg) in THF cooled at -60 ℃. After transfer completion, the resulting mixture was warmed to room temperature and stirred for 1 h. The reaction was quenched with water, extracted with petroleum. The organic phase was washed with brine, dried over sodium sulfate, condensed to give 25.1 Kg of Compound-3. 1H NMR (400 MHz, DMSO-d6) δ 3.34 (d, J = 16.0 Hz, 1H) , 3.15 (m, 1H) , 2.78 (d, J = 16.0 Hz, 1H) , 2.27 (m, 1H) , 1.93 (m, 1H) , 1.68 (m, 3H) , 1.41 (s, 9H) , 1.24 (s, 3H) , 0.13 (s, 9 H) ppm.
Step 3: Synthesis of Compound-4

To a cool (0-5 ℃) solution of 70.1 Kg of Compound-3 in THF was added tetrabutylammonium fluoride (13.3 Kg) in THF. After de-silylation was completed, the reaction was quenched with water, extracted with petroleum (290 Kg) and the organic phase was condensed and passed through a pad of silica gel. The filtrate was condensed to give 48 Kg of Compound-4. 1H NMR (400 MHz, DMSO-d6) δ 3.36 (d, J = 16.0 Hz, 1H) , 3.15 (m, 1H) , 2.82 (d, J = 16.0 Hz, 1H) , 2.28 (m, 1H) , 1.97 (m, 1H) , 1.70 (m, 3H) , 1.41 (s, 9H) , 1.26 (s, 3H) ppm.
Step 4: Syntheses of Compound-5

A solution of Compound-4 (48 Kg) in THF was warmed to 50-60 ℃. To the above solution was added a solution of (-) -di-p-methylbenzoyl-L-tartaric acid (69.6 Kg) in THF. The resulting mixture was stirred at 50-60 ℃ 1-2 h and then gradually cooled to 0-10 ℃. The resulting salt solid was filtered and re-suspended in methyl tert-butyl ether and heated at 50-60 ℃ for 1 h. The mixture was gradually cooled to 0-5 ℃. The resulting solid was filtered to give 13.1 Kg of off-white solid. The solid was treated with aqueous sodium hydroxide, extracted with petroleum, condensed to give 13.1 Kg of Compound-5 (ee≥96%) . 1H NMR (400 MHz, DMSO-d6) δ 3.36 (d, J = 16.0 Hz, 1H) , 3.15 (m, 1H) , 2.82 (d, J = 16.0 Hz, 1H) , 2.29 (m, 1H) , 1.97 (m, 1H) , 1.70 (m, 3H) , 1.41 (s, 9H) , 1.26 (s, 3H) ppm.
Step 5: Syntheses of Compound-6

Intermediate B (14 Kg) , bis (triphenyl) palladium dichloride (0.7 Kg) , CuI (0.42 Kg) and tetramethyl guanidine (11.5 Kg) were dissolved in DMF (48.1 Kg) . The resulting solution was stirred and de-gassed and then heated under nitrogen. A solution of Compound-5 (9.24 Kg) in DMF (16 Kg) was added dropwise. After coupling, the organic phase was condensed, the resiue was stirred with water (145 Kg) and methyl t-butyl ether (104 Kg) , the entire mixture passed trough a pad of celite, separated. The organic phase was washed with a solution of thiourea (14 Kg) in water (165 kg) and brine (100 Kg) , condensed. The residue was dissolved in a mixture of n-heptane (120 Kg) and ethyl acetate (28 Kg) . The solution was mixed with charcoal (1.4 kg) , heated at 40-50 ℃ for 1-2 h, fltered though a pad of silica gel. The filtrate was condensed to give Compound-6 solid (14.89 Kg) and the liquid filtrate (13 Kg heptane solution, contains 1.24 Kg of Compound-6) . 1H NMR (400 MHz, DMSO-d6) δ 7.85 (d, J = 9.6 Hz, 1H) , 7.55 (m, 3H) , 7.32 (m, 2H) , 3.87 (s, 3H) , 3.37 (d, J = 16.0 Hz, 1H) , 3.22 (m , 1H) , 2.94 (d, J = 16.0, Hz, 1H) , 2.60 (m, 1H) , 2.48 (m, 1H) , 2.29 (s, 3h) , 2.26 (m, 1 H) , 1.82 (m, 2H) , 1.49 (s, 3H) , 1.43 (s, 9H) ppm.
Step 6: Syntheses of Compound-7

The above heptane solution of Compound-6 was added into a cold trifluoromethane sulfonic acid (66.1 Kg) while maintaining the internal temperature below 25 ℃. Then solid Compound-6 (14.87 Kg) was added batchwise. After complete addition of Compound-6, the reaction mixture was warmed to 25-30℃ and stiired until the reaction was completed. The entire mixture was poured into a solution of sodium acetate (123.5 Kg) in water (240 Kg) . pH of the solution was then adjusted to 7-8 by adding solid potassium carbonate (46.1 Kg) . The mixture was extracted wuth dichloromethane (509 Kg) , condensed. The residue was mixed with n-heptane (41 Kg) , condensed again to give the precipitate which was filtered and washed by n-heptane (8 Kg) and dried. 8.78 Kg of Compound-7 was obtained. 1H NMR (400 MHz, DMSO-d6) δ 12.30 (s, 1H) , 7.35 (dd, J = 9.2, 1.6 Hz, 1H) , 7.08 (dd, J = 9.2, 1.6 Hz, 1H) , 3.79 (s, 3H) , 3.68 (d, J = 17.2 Hz, 1H) , 3.21 (d, J = 17.2 Hz, 1H) , 3.06 (m, 1H) , 2.68 (m, 1H) , 1.96 (m, 1H) , 1.74 (m, 1H) , 1.49 (s, 3H) ppm.
Step 7: Syntheses of Compound 1 –Crude 1

Compound-7 (8.76 Kg) was dissolved in methanol (69 Kg) and internally cooled below 25 ℃. Acetic acid (9.3 Kg) and hydrazine hydrate (7.4 Kg, 85%) were added while maintaining internal temperature below 25 ℃. After de-gassed and re-filled with nitrogen (repeated three times) , the reaction mixture was stirred at 55-60 ℃ for 4 h. After a complete reaction, the mixture was mixed with water (29 Kg) . The organic phase was condensed and potassium carbonate (12.5 Kg) in water (40 Kg) was added. The resulting solid was filtered, washed with water (18.3 Kg) . The solid was slurred with water (110 Kg) , centrifuged, dried and slurred with ethanol (9.4 Kg) , centrifuged, filtered, washed with ethanol, dried in vacuum to give Compound 1-Crude 1 (7.91 Kg) . 1H-NMR (600 MHz, DMSO-d 6) δ 12.0 (s, 1H) , 10.2 (s, 1H) , 7.31 (dd, 1H, J=9.6, 2.0 Hz) , 7.19 (dd, 1H, J=9.6, 2.0 Hz) , 3.77 (d, 1H, J=16.4 Hz) , 3.34 (d, 1H, J=16.4 Hz) , 2.97-3.02 (m, 1H) , 2.54-2.58 (m, 1H) , 2.35-2.40 (m, 1H) , 1.90-1.94 (m, 1H) , 1.73-1.75 (m, 1H) , 1.47 (s, 3H) , 1.43-1.45 (m, 1H) ppm. MS (ESI) m/e [M+1] + 299.
Step 8: Synthesis of Compound 1-Crude 2

Under nitrogen protection, Compound 1 (Crude 1) (7.88 Kg) was stirred with isopropanol (422 Kg) and heated at 70-80 ℃ for 1-2 h until the solid disappeared completely. A solution of (+) -di-p-methylbenzoyl-D-tartaric acid (10.25 Kg) in isopropanol (84.4 Kg) was added. The mixture was stirred for 14-16 h, filtered and washed with isopropanol (16 Kg) , dried. The resulting salt was added into a stirred solution of potassium carbonate (6.15 Kg) in water (118 Kg) . The precipitate was centrifuged, filtered, washed with water (18 Kg) . The solid was slurred with water (110 Kg) , centrifuged, dried. The solid was dissolved in THF (75 Kg) , active carbon (0.8 Kg) was added. The mixture was degassed and re-protected by nitrogen, stirred and heated at 40-45 ℃ for 1-2 h, cooled, filtered through celite, condensed to give the solid which was further slurred with ethanol (6.5 Kg) , filtered to give 5.6 Kg of Compound
1 crude
2. 1H NMR (400 MHz, DMSO-d6) δ 12.0 (s, 1H) , 10.2 (s, 1H) , 7.31 (dd, 1H, J=9.6, 2.0 Hz) , 7.19 (dd, 1H, J=9.6, 2.0 Hz) , 3.77 (d, 1H, J=16.4 Hz) , 3.34 (d, 1H, J=16.4 Hz) , 2.97-3.02 (m, 1H) , 2.54-2.58 (m, 1H) , 2.35-2.40 (m, 1H) , 1.90-1.94 (m, 1H) , 1.73-1.75 (m, 1H) , 1.47 (s, 3H) , 1.43-1.45 (m, 1H) ppm. MS (ESI) m/e [M+1] + 299.
PATENT
WO 2017032289
https://patents.google.com/patent/WO2017032289A1/en
Scheme 1: Synthetic Process of Compound A in a large scale

PATENT
WO 2013097225
https://patents.google.com/patent/WO2013097225A1/en
Example 36: Synthesis of Compound 69 Compound 69: (RV2-fluoro-10a-methyl-7,8,9 JO .10a.l l-hexahydro-5,6,7a,l 1- tetraazacvcloheptardeflcyclopentara1fluoren-4(5H)-one

Step 1 : Methyl 2-bromo-5-fluoro-3-(2,2,2-trifluoroacetamido)benzoate

To a solution of methyl 3-amino-2-bromo-5-fluorobenzoate (25. Og, 100 mmol) and K2CO3 (42.0g, 302 mmol) in DCM (250mL) were added 2,2,2-trifluoroacetic anhydride (249.0g, 1.197mol) at 5 -10°C under nitrogen atmosphere. The mixture was stirred for overnight at 25°C. The reaction mixture was diluted with DCM, washed with H20 (200mLx2) and saturared
NaHCC”3 aq (200mLx2), dried over anhydrousNa2S04, and concentrated to give 34.0 g (98%) of methyl 2-bromo-5-fluoro-3-(2,2,2-trifluoroacetamido)benzoate as white solid. 1H NMR (CDCI3– dl) δ 8.87 (s, 1H), 8.36 (d, 1H,J=6.4 Hz), 7.43 (d, 1H,J=5.2 Hz), 3.98 (s, 3H).
Step 2: (R)-benzyl 2-((4-fluoro-2-(methoxycarbonyl)-6- (2,2,2trifluoroacetamido)phenyl)ethvnyl)-2-methylpyrrolidine-l-carboxylate

A mixture of methyl 2-bromo-5-fluoro-3-(2,2,2-trifluoroacetamido)benzoate (27.52g, 80 mmol), (PPh3)2PdCl2 (2.8 g, 4 mmol), (R)-benzyl 2-ethynyl-2-methylpyrrolidine-l-carboxylate (19.44 g, 80 mmol),copper(I) iodide (764 mg, 4 mmol) and tetramethylguanidine (27.6 g, 240 mmol) in DMF (200 mL) was heated at 80 °C with nitrogen protection system for 16 hours. The cooled reaction mixture was diluted with EA (3×200 mL) and water (800 mL). The organic layer was separated, washed with water (2×200 mL), dried (Na2S04), and concentrated. The remaining residue was chromatographed on silica gel, eluted with gradient 0-30% EtOAc in hexane to give the product (R)-benzyl 2-((4-fluoro-2-(methoxycarbonyl)-6-
(2,2,2trifluoroacetamido)phenyl)ethynyl)-2-methylpyrrolidine-l-carboxylate (21 g, 53%) as white solid. 1H NMR (DMSO-dl) δ 11.01 (s, 1H), 7.64-7.77 (m, 1H), 7.36 (m, 5H),7.19-7.31 (m, 1H), 5.04-5.12 (m, 2H), 3.85(s, 3H ), 3.44-3.47 (m, 2H), 2.0-2.29 (m, 2H), 1.90-1.97 (m, 2H), and 1.69 (s, 3H).MS (ESI) m/e [M+l]+ 507.0.
Step 3: (R)-methyl 6-fluoro-2-(2 -methyl- l-(2,2,2-trifluoroacetyl)pyrrolidin-2-yl)-lH-indole-4- carboxylate

To a solution of (R)-benzyl 2-((4-fluoro-2-(methoxycarbonyl)-6- (2,2,2trifiuoroacetamido)phenyl)ethynyl)-2-methylpyrrolidine- 1 -carboxylate(5.0g, 1 Ommol) in toluene was added zinc(II) bromide(l 1.25g, 50 mmol) at room temperture. The reaction mixture was heated at 80 °C with nitrogen protection system for 15 hours. The solvent was removed under reduced pressure, and the residue was treated with DCM (500 mL) and water (800 mL). The organic layer was separated, washed with water (2×200 mL), dried (Na2S04), and
concentrated. The remaining residue was chromatographed on silica gel ,eluted with gradient 0- 50% EtOAc in hexane to give the product(R)-methyl 6-fluoro-2-(2 -methyl- 1 -(2,2,2- trifluoroacetyl)pyrrolidin-2-yl)-lH-indole-4-carboxylate (1.9 g, 51%) as yellow solid. 1H NMR (CDCls-dl) δ 9.97 (s, 1H), 7.62 (d,lH, J=10.2 Hz), 7.27 (d,lH, J=9.6 Hz), 7.05 (d,lH, J=1.2 Hz), 3.98 (s, 3H), 3.86-3.88 (m,2H),2.91-2.96 (m,lH), 2.25-2.28 (m,lH), 2.12-2.16 (m, 2H), and 1.99 (s, 3H). MS (ESI) m/e [M+l]+ 507.0.
Step 4: (R)-methyl 6-fluoro-2-(2-methylpyrrolidin-2-yl)-lH-indole-4-carboxylate

To a solution of (R)-methyl 6-fluoro-2-(2 -methyl- l-(2,2,2-trifluoroacetyl)pyrrolidin-2-yl)- lH-indole-4-carboxylate (1.0 g, 1.9 mmol) in MeOH was added NaBH4 (706 mg, 11.4 mmol) at room temperature. The reaction mixture was refluxed for 4 hours with nitrogen protection system. The solvent was removed under reduced pressure. The residue was dissolved in DCM (200 mL), which was washed with water (200 mL)and brine (200 mL), dried over Na2S04, and concentrated to give the desire product as yellow oil. (R)-methyl 6-fluoro-2-(2-methylpyrrolidin- 2-yl)-lH-indole-4-carboxylate (727 mg, 98%). 1H NMR (CD3OD-dl) δ 7.50(dd,lH, J=10.2, 2.4 Hz), 7.32 (d,lH, J=9.0, 2.4 Hz), 6.93 (s, 1H),3.97 (s, 3H), 3.03-3.12 (m, 2H), 2.27-2.32 (m, 1H),1.88-1.98 (m, 3H), and 1.60 (s, 3H). MS (ESI) m/e [M+l]+ 276.0.
Step 5: (R)-Methyl 6-fluoro-2-(l-(2-methoxy-2-oxoethyl)-2-methylpyrrolidin-2-yl)-lH-indole-4- carboxylate

To a stirred mixture of (R)-methyl 6-fluoro-2-(2-methylpyrrolidin-2-yl)-lH-indole-4- carboxylate (1.0, 1.27 mol), CH3CN (50 ml) and methylbromoacetate (0.58 g, 3.82mmol) was added DIPEA(0.82 g, 6.35 mmol). The reaction mixture was stirred at room temperature for about 20 hours. The reaction mixture was then diluted with CH2CI2 (15 ml) and washed with water three times. The organic layer was dried with MgS04 and concentrated to give 0.85 g of (R)-methyl 6-fluoro-2-(l-(2-methoxy-2-oxoethyl)-2-methylpyrrolidin-2-yl)-lH-indole-4- carboxylate. 1H NMR (CD3OD-d4) δ 7.47 (dd, 1H, J=2.4, 12.0 Hz), 7.27 (dd, 1H, J=2.4, 9.0 Hz), 6.89 (s,lH), 3.95 (s, 3H), 3.66-3.68 (m, 1H), 3.64 (s, 3H), 3.16-3.17 (m, 2H), 2.72-2.75 (m, 1H), 1.88-2.02 (m, 4H), and 1.44 (s, 3H).MS (ESI) m/e [M+l]+ 349.0.
Step 6: (R)-methyl 9-fluoro-l lb-methyl-6-oxo-2,3, 5,6, 11,1 lb-hexahydro-lH-indolizinor8,7- blindole-7-carboxylate

In a 25 -mL flask, (R)-methyl 6-fluoro-2-(l-(2-methoxy-2-oxoethyl)-2-methylpyrrolidin-2- yl)-lH-indole-4-carboxylate (100 mg) was treated with anhydrous MeS03H (6 mL). The flask was fitted with a reflux condenser and heated at 60 °C for 1 h. Then, the reaction mixture was cooled in an ice-bath and diluted with distilled water (6.0 mL). The pH of the solution was increased to pH~10 by the addition of saturated aq. NaHC03. The reaction mixture was then extracted with EtOAc (3×5 mL). Theorganic extracts were combined and washed with brine (lx5mL), dried over Na2S04, filtered, and concentrated. The residue was purified by Pre-TLC to give (R)-methyl 9-fluoro-l lb-methyl-6-oxo-2,3, 5,6,11,1 lb-hexahydro-lH-indolizino[8,7- b]indole-7-carboxylate(30 mg). 1H NMR (CDCl3-d) δ 7.14-7.224 (m, 2H), 4.03 (s, 3H), 3.81- 3.84 (m, 1H), 3.57-3.59 (m, 1H), 3.22-3.24 (m, 1H), 2.92-2.94 (m, 1H), 2.39-2.40 (m,lH), 2.16- 2.17 (m,lH),1.93-1.94 (m, 1H), 1.63 (s, 3H), and 1.56-1.57 (m, 1H).MS (ESI) m e [M+l]+ 317.0. Step 7: (RV2-fluoro-10a-methyl-7,8,9 JO JOa.l l-hexahvdro-5.6.7a.l 1- tetraazacyclohepta[def|cyclopenta[alfluoren-4(5H)-one

A solution of compound (R)-methyl 9-fluoro-l lb-methyl-6-oxo-2,3,5,6,l 1,1 lb-hexahydro- lH-indolizino[8,7-b]indole-7-carboxylate (90 mg), acetic acid (0.54 g), and hydrazine hydrate (0.28g) in methanol (30 mL) was heated at reflux. After 5 h, the reaction was cooled and water (5 mL) was added.The mixture was extracted with EtOAc (3×5 mL). The combined organic layers were washed with brine (10 mL) and driedover MgSC^. The mixture was filtered, and the filtrate was evaporated to dryness, and the residue was purified by Pre-TLC using CH2CI2 as eluent to give 80 mg of (R)-2-fluoro-10a-methyl-7,8,9,10,10a,l l-hexahydro-5,6,7a,l l- tetraazacyclohepta[defJcyclopenta[a]fluoren-4(5H)-one. 1H NMR (DMSO-d6) δ 11.9 (s, 1H), 10.2 (s, 1H), 7.30 (d, 1H, J=9.6 Hz), 7.20 (d, 1H, J=10.2 Hz), 3.76 (d, 1H, J=16.4 Hz), 3.34 (d, 1H, J=16.4 Hz), 2.99-3.02 (m, 1H), 2.54-2.58 (m, 1H), 2.35-2.40 (m, 1H), 1.90-1.94 (m, 1H), 1.73-1.75 (m, 1H), 1.48 (s, 3H), and 1.43-1.45(m, 1H). MS (ESI) m/e [M+l]+ 299.
SYN
https://doi.org/10.1021/acs.jmedchem.3c02374
J. Med. Chem. 2024, 67, 4376−4418
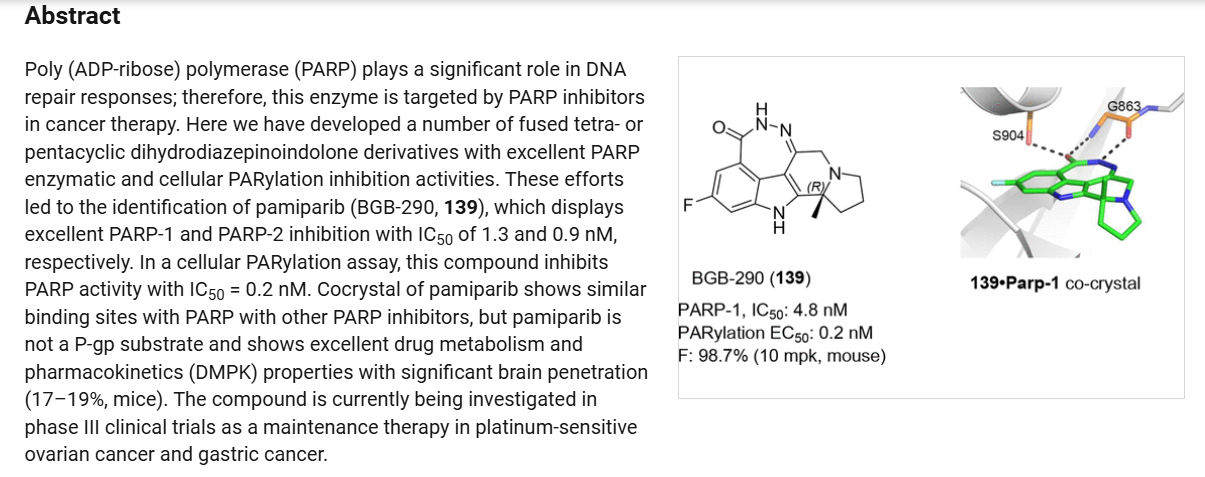
Pamiparib (Partruvix). Pamiparib (27) is an orally active, potent, and highly selective PARP1 and PARP2
inhibitor being developed by BeiGene Limited.190 The drug was approved in China in 2022 for the treatment of germline BRCA-mutated recurrent advanced ovarian, fallopian tube, or primary peritoneal cancer.190BRCA1 and BRCA2 are critical tumor suppressors that help DNA double-strand break (DSB)
repair by functional homologous recombination (HR). 191,192 192 It was claimed that pamiparib showed good brain penetration ability for the treatment of cancer patients with brain metastasis. A small-scale synthesis of pamiparib (27) was first disclosed by BeiGene Limited in 2013.193 Later, they reported a
modified route for industrial scale preparation of the API which is described below. 194,195
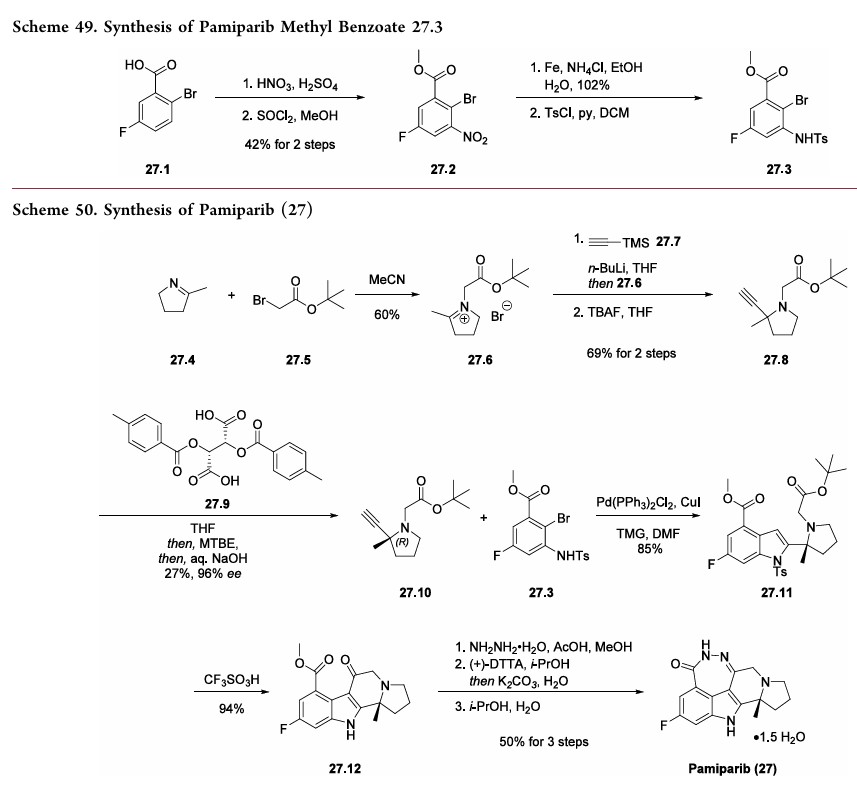
The synthesis commenced with 2-bromo-5-fluorobenzoic acid (27.1) which was subjected to nitration followed by esterification to deliver methyl benzoate 27.2 in 42% overall yield (Scheme 49). The nitro
derivative 27.2 was reduced to an aniline and subsequently protected as a tosylate to obtain the key aryl bromide fragment 27.3. It should be noted that a yield for the tosylation step was not provided by the inventors.
Preparation of the other key fragment 27.10 and endgame of the pamiparib synthesis are described in Scheme 50. First,pyrroline 27.4 was treated with t-butyl bromoacetate 27.5 to generate iminium bromide salt 27.6. An acetylide derived from trimethylsilyl acetylene 27.7 was then added to the iminium to
install the tertiary center. A TBAF-mediated removal of the silyl moiety delivered racemic alkyne 27.8 in 69% yield over two steps. The enantiomers were separated via a classical salt resolution with (−)-di-p-methylbenzolyl-L-tartaric acid (27.9). The desired (R)-enantiomer 27.10 was obtained in 96%
enantiomeric excess (ee) after isolation as the free-base amine. The authors explored several routes to access 27.10; however,the salt resolution approach was selected due to its scalability and reproducibility.
192With the alkyne subunit 27.10 and bromide subunit 27.3 in hand, the next objective was combining them in a convergent manner. This was achieved via an efficient Larock heteroannulation reaction, affording indole 27.11 in 85% yield. Treatment of diester 27.11 with triflic acid triggered removal of both t-butyl ester and N-tosyl protecting groups, as well as cyclization to generate tetracycle 27.12 in 94% yield. The ketoester 27.12 was subjected to hydrazine hydrate in the presence of acetic acid to deliver the
crude cyclized material which was purified via salt formation with (+)-DTTA. Finally, treatment of the amine precursor with water in hot isopropanol delivered pamiparib (27) as a sesquihydrate crystalline solid in 50% over 3 steps.
(190) Markham, A. Pamiparib: First approval. Drugs 2021, 81,1343−1348.
(191) Xiong, Y.; Guo, Y.; Liu, Y.; Wang, H.; Gong, W.; Liu, Y.;Wang, X.; Gao, Y.; Yu, F.; Su, D.; et al. Pamiparib is a potent andselective PARP inhibitor with unique potential for the treatment of brain tumor. Neoplasia 2020, 22, 431−440.
(192) Wang, H.; Ren, B.; Liu, Y.; Jiang, B.; Guo, Y.; Wei, M.; Luo,L.; Kuang, X.; Qiu, M.; Lv, L.; et al. Discovery of pamiparib (BGB290), a potent and selective poly (ADP-ribose) polymerase (PARP)inhibitor in clinical development. J. Med. Chem. 2020, 63, 15541−15563.
(193) Zhou, C.; Ren, B.; Wang, H. Fused tetracyclic and pentacyclic
dihydrodiazepinocarbazolones as PARP inhibitors and their prepara
tion. WO 2013097225 A1, 2013.
(194) Wang, H.; Zhou, C.; Ren, B.; Kuang, X. Process for preparing
(R)-2-fluoro-10a-methyl-7,8,9,10,10a,11-hexahydro-5,6,7a,11
tetraazacyclohepta[def]cyclopenta[a]fluoren-4(5H)-one as PARP in
hibitor, crystalline forms, and uses thereof. WO 2017032289 A1,
2017.
(195) Wang, H.; Kuang, X.; Zhou, C. Crystalline forms of salts of
fused tetra or penta-cyclic dihydrodiazepinocarazolones, and uses
thereof. WO 2018157794 A1, 2018.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- Xiong Y, Guo Y, Liu Y, Wang H, Gong W, Liu Y, et al. (September 2020). “Pamiparib is a potent and selective PARP inhibitor with unique potential for the treatment of brain tumor”. Neoplasia. 22 (9): 431–440. doi:10.1016/j.neo.2020.06.009. PMC 7350150. PMID 32652442.
- Markham A (July 2021). “Pamiparib: First Approval”. Drugs. 81 (11): 1343–1348. doi:10.1007/s40265-021-01552-8. PMID 34287805.
- Friedlander M, Mileshkin L, Lombard J, Frentzas S, Gao B, Wilson M, et al. (September 2023). “Pamiparib in combination with tislelizumab in patients with advanced solid tumours: results from the dose-expansion stage of a multicentre, open-label, phase I trial”. British Journal of Cancer. 129 (5): 797–810. doi:10.1038/s41416-023-02349-0. PMC 10449784. PMID 37474720.
| Clinical data | |
|---|---|
| Trade names | Partruvix |
| Other names | BGB-290 |
| ATC code | L01XK06 (WHO) |
| Legal status | |
| Legal status | US: Investigational New DrugRx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1446261-44-4 |
| PubChem CID | 135565554 |
| DrugBank | DB14769 |
| ChemSpider | 58805610 |
| UNII | 8375F9S90C |
| KEGG | D11426 |
| ChEMBL | ChEMBL4112930 |
| Chemical and physical data | |
| Formula | C16H15FN4O |
| Molar mass | 298.321 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
{kind=link}
/////////////Pamiparib, APPROVALS 2022, CHINA 2022, BeiGene, BGB 290