















Plinabulin
- Molecular FormulaC19H20N4O2
- Average mass336.388 Da
Tubulin antagonist
Cancer; Febrile neutropenia; Non-small-cell lung cancer
Plinabulin (chemical structure, BPI-2358, formerly NPI-2358) is a small molecule under development by BeyondSpring Pharmaceuticals, and is in a world-wide Phase 3 clinical trial for non-small cell lung cancer. [1] Plinabulin blocks the polymerization of tubulin in a unique manner, resulting in multi-factorial effects including an enhanced immune-oncology response, [2] activation of the JNK pathway [3] and disruption of the tumor blood supply. Plinabulin is being investigated for the reduction of chemotherapy-induced neutropenia [4] and for anti-cancer effects in combination with immune checkpoint inhibitors [5] [6] and in KRAS mutated tumors. [7]
Plinabulin is a synthetic analog of diketopiperazine phenylahistin (halimide) discovered from marine and terrestrial Aspergillus sp. Plinabulin is structurally different from colchicine and its combretastatin-like analogs (eg, fosbretabulin) and binds at or near the colchicine binding site on tubulin monomers. Previous studies showed that plinabulin induced vascular endothelial cell tubulin depolymerization and monolayer permeability at low concentrations compared with colchicine and that it induced apoptosis in Jurkat leukemia cells. Studies of plinabulin as a single agent in patients with advanced malignancies (lung, prostate, and colon cancers) showed a favorable pharmacokinetic, pharmacodynamics, and safety profile.
Beyondspring, under license from Nereus (now Triphase, which licensed the program from the Scripps Institute of Oceanography of the University of California San Diego), is developing plinabulin, the lead in the NPI-2350 halimide series of marine Aspergillus-derived, vascular-targeting antimicrotubule agents, for treating cancer, primarily non-small cell lung cancer.

It is thought that a single, universal cellular mechanism controls the regulation of the eukaryotic cell cycle process. See, e.g., Hartwpll, L.H. et al., Science (1989), 246: 629-34. It is also known that when an abnormality arises in the control mechanism of the cell cycle, cancer or an immune disorder may occur. Accordingly, as is also known, antitumor agents and immune suppressors may be among the substances that regulate the cell cycle. Thus, new methods for producing eukaryotic cell cycle inhibitors are needed as antitumor and immune-enhancing compounds, and should be useful in the treatment of human cancer as chemotherapeutic, anti-tumor agents. See, e.g., Roberge, M. et al., Cancer Res. (1994), 54, 6115-21.
Fungi, especially pathogenic fungi and related infections, represent an increasing clinical challenge. Existing antifungal agents are of limited efficacy and toxicity, and the development and/or discovery of strains of pathogenic fungi that are resistant to drags currently available or under development. By way of example, fungi that are pathogenic in humans include among others Candida spp. including C. albicans, C. tropicalis, C. keƒyr, C. krusei and C. galbrata; Aspergillus spp. including A. fumigatus and A. flavus; Cryptococcus neoƒormans; Blastomyces spp. including Blastomyces dermatitidis; Pneumocystis carinii; Coccidioides immitis; Basidiobolus ranarum; Conidiobolus spp.; Histoplasma capsulatum; Rhizopus spp. including R. oryzae and R. microsporus; Cunninghamella spp.; Rhizomucor spp.; Paracoccidioides brasiliensis; Pseudallescheria boydii; Rhinosporidium seeberi; and Sporothrix schenckii (Kwon-Chung, K.J. & Bennett, J.E. 1992 Medical Mycology, Lea and Febiger, Malvern, PA).
Recently, it has been reported that tryprostatins A and B (which are diketopiperazines consisting of proline and isoprenylated tryptophan residues), and five other structurally-related diketopiperazines, inhibited cell cycle progression in the M phase, see Cui, C. et al., 1996 J Antibiotics 49:527-33; Cui, C. et al. 1996 J Antibiotics 49:534-40, and that these compounds also affect the microtubule assembly, see Usui, T. et al. 1998 Biochem J 333:543-48; Kondon, M. et al. 1998 J Antibiotics 51:801-04. Furthermore, natural and synthetic compounds have been reported to inhibit mitosis, thus inhibit the eukaryotic cell cycle, by binding to the colchicine binding-site (CLC-site) on tubulin, which is a macromolecule that consists of two 50 kDa subunits (α- and β-tubulin) and is the major constituent of microtubules. See, e.g., Iwasaki, S., 1993 Med Res Rev 13:183-198; Hamel, E. 1996 Med Res Rev 16:207-31; Weisenberg, R.C. et al., 1969 Biochemistry 7:4466-79. Microtubules are thought to be involved in several essential cell functions, such as axonal transport, cell motility and determination of cell morphology. Therefore, inhibitors of microtubule function may have broad biological activity, and be applicable to medicinal and agrochemical purposes. It is also possible that colchicine (CLC)-site ligands such as CLC, steganacin, see Kupchan, S.M. et al., 1973 J Am Chem Soc 95:1335-36, podophyllotoxin, see Sackett, D.L., 1993 Pharmacol Ther 59:163-228, and combretastatins, see Pettit, G.R. et al., 1995 J Med Chem 38:166-67, may prove to be valuable as eukaryotic cell cycle inhibitors and, thus, may be useful as chemotherapeutic agents.
Although diketopiperazine-type metabolites have been isolated from various fungi as mycotoxins, see Horak R.M. et al., 1981 JCS Chem Comm 1265-67; Ali M. et al., 1898 Toxicology Letters 48:235-41, or as secondary metabolites, see Smedsgaard J. et al., 1996 J Microbiol Meth 25:5-17, little is known about the specific structure of the diketopiperazine-type metabolites or their derivatives and their antitumor activity, particularly in vivo. Not only have these compounds been isolated as mycotoxins, the chemical synthesis of one type of diketopiperazine-type metabolite, phenylahistin, has been described by Hayashi et al. in J. Org. Chem. (2000) 65, page 8402. In the art, one such diketopiperazine-type metabolite derivative, dehydrophenylahistin, has been prepared by enzymatic dehydrogenation of its parent phenylahistin. With the incidences of cancer on the rise, there exists a particular need for chemically producing a class of substantially purified diketopiperazine-type metabolite-derivatives having animal cell-specific proliferation-inhibiting activity and high antitumor activity and selectivity. There is therefore a particular need for an efficient method of synthetically producing substantially purified, and structurally and biologically characterized, diketopiperazine-type metabolite-derivatives.
Also, PCT Publication WO/0153290 (July 26, 2001) describes a non-synthetic method of producing dehydrophenylahistin by exposing phenylahistin or a particular phenylahistin analog to a dehydrogenase obtained from Streptomyces albulus.
Synthesis


PATENT
WO2001053290,
WO 2004054498
PATENT
The imidazolecarboxaldehyde may be prepared, for example, according the procedure disclosed in Hayashi et al., 2000 J Organic Chem 65: 8402 as depicted below:

EXAMPLE 2
Synthesis and Physical Characterization of tBu-dehydrophenylahistin Derivatives
[0207] Structural derivatives of dehydrophenylahistin were synthesized according to the following reaction schemes to produce tBu-dehydrophenylahistin. Synthesis by Route
A (see Figure 1) is similar in certain respects to the synthesis of the dehydrophenylahistin synthesized as in Example 1.


Route A:
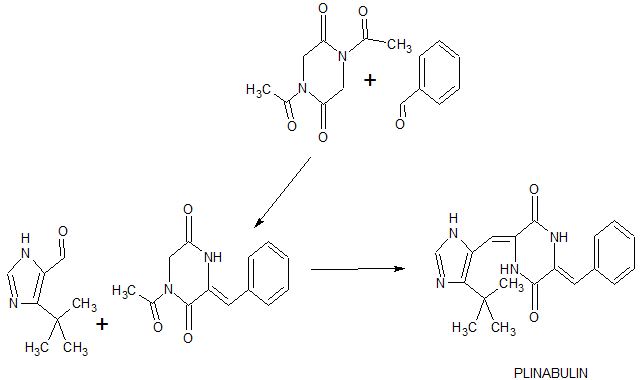
[0208] N,N’-diacethyl-2,5-piperazinedione 1 was prepared as in Example 1.
1) 1-Acetyl-3-{(Z)-1-[5-tert-butyl-1H-4-imidazolyl]methylidene}]-2,5-piperazinedione (16)

. [0209] To a solution of 5-tert-butylimidazole-4-carboxaldehyde 15 (3.02 g, 19.8. mmol) in DMF (30 mL) was added compound 1 (5.89 g, 29.72 mmol) and the solution was repeatedly evacuated in a short time to remove oxygen and flushed with Ar, followed by the addition of Cs2CO3 (9.7 g, 29.72 mmol) and the evacuation-flushing process was repeated again. The resultant mixture was stirred for 5 h at room temperature. After the solvent was removed by evaporation, the residue was dissolved in the mixture of EtOAc and 10% Na2CO3, and the organic phase was washed with 10% Na2CO3 again and saturated NaCl for three times, dried over Na2SO4 and concentrated in vacuo. The residual oil was purified by column chromatography on silica using CHCl3-MeOH (100:0 to 50:1) as an eluant to give 1.90 g (33 %) of a pale yellow solid 16. 1H NMR (270 MHz, CDCl3) δ 12.14 (d, br-s, 1H), 9.22 (br-s, 1H), 7.57 (s, 1H), 7.18, (s, 1H), 4.47 (s, 2H), 2.65 (s, 3H), 1.47 (s, 9H).
2) t-Bu-dehydrophenylahistin

[0210] To a solution of 1-Acetyl-3-{(Z)-1-[5-tert-butyl-1H-4-imidazolyl]methylidene}]-2,5-piperazinedione (16) (11 mg, 0.038 mmol) in DMF (1.0 mL) was added benzaldehyde (19 μL, 0.19 mmol, 5 eq) and the solution was repeatedly evacuated in a short time to remove oxygen and flushed with Ar, followed by the addition of Cs2CO3 (43 mg, 0.132 mmol, 3.5 eq) and the evacuation-flushing process was repeated again. The resultant mixture was heated for 2.5 h at 80°C. After the solvent was removed by
evaporation, the residue was dissolved in EtOAc, washed with water for two times and saturated NaCl for three times, dried over Na2SO4 and concentrated in vacuo. The resulting residue was dissolved in 90% MeOH aq and applied to reverse-phase HPLC column (YMC-Pack, ODS-AM, 20 × 250 mm) and eluted using a linear gradient from 70 to 74% MeOH in water over 16 min at a flow rate of 12 mL/min, and the desired fraction was collected and concentrated by evaporation to give a 6.4 mg (50%) of yellow colored tert-butyl-dehydrophenylahistin. 1H NMR (270 MHz, CDCl3) δ 12.34 br-s, 1H), 9.18 (br-s, 1H), 8.09 (s, 1H), 7.59 (s, 1H), 7.31 – 7.49 (m, 5H), 7.01 s, 2H), 1.46 (s, 9H).
[0211] The dehydrophenylahistin reaction to produce tBu-dehydrophenylahistin is identical to Example 1.
[0212] The total yield of the tBu-dehydrophenylahistin recovered was 16.5%. Route B:
[0213] N,N’-diacethyl-2,5-piperazinedione 1 was prepared as in Example 1.
1) 1-Acetyl-3-[(Z)-benzylidenel]-2,5-piperazinedione (17)

[0214] To a solution of benzaldehyde 4 (0.54 g, 5.05. mmol) in DMF (5 mL) was added compound 1 (2.0 g, 10.1 mmol) and the solution was repeatedly evacuated in a short time to remove oxygen and flushed with Ar, followed by the addition of Cs2CO3 (1.65 g, 5.05 mmol) and the evacuation-flushing process was repeated again. The resultant mixture was stirred for 3.5 h at room temperature. After the solvent was removed by evaporation, the residue was dissolved in the mixture of EtOAc and 10% Na2CO3, and the organic phase was washed with 10% Na2CO3 again and saturated NaCl for three times, dried over Na2SO4 and concentrated in vacuo. The residual solid was recrystalized from MeOH-ether to obtain a off-white solid of 17; yield 1.95 g (79%).
2) t-Bu-dehydrophenylahistin

[0215] To a solution of 1-Acetyl-3-[(Z)-benzylidenel]-2,5-piperazinedione (17) (48 mg, 0.197 mmol) in DMF (1.0 mL) was added 5-tert-butylimidazole-4-carboxaldehyde 15 (30 mg, 0.197 mmol) and the solution was repeatedly evacuated in a short time to remove oxygen and flushed with Ar, followed by the addition of Cs2CO3 (96 mg, 0.296 mmol) and the evacuation-flushing process was repeated again. The resultant mixture was heated for 14 h at 80°C. After the solvent was removed by evaporation, the residue was dissolved in EtOAc, washed with water for two times and saturated NaCl for three times, dried over Na2SO4 and concentrated in vacuo. The resulting residue was dissolved in 90% MeOH aq and applied to reverse-phase HPLC column (YMC-Pack, ODS-AM, 20 x 250 mm) and eluted using a linear gradient from 70 to 74% MeOH in water over 16 min at a flow rate of 12 mL/min, and the desired fraction was collected and concentrated by evaporation to give a 0.8 mg (1.2%) of yellow colored tert-butyl-dehydrophenylahistin.
[0216] The total yield of the tBu-dehydrophenylahistin recovered was 0.9%.
[0217] The HPLC profile of the crude synthetic tBu-dehyrophenylahistin from Route A and from Route B is depicted in Figure 4.
[0218] Two other tBu-dehydrophenylahistin derivatives were synthesized according to the method of Route A. In the synthesis of the additional tBu-dehydrophenylahistin derivatives, modifications to the benzaldehyde compound 4 were made.
[0219] Figure 4 illustrates the similarities of the HPLC profiles (Column: YMC-Pack ODS-AM (20 × 250mm); Gradient: 65% to 75% in a methanol-water system for 20 min, then 10 min in a 100% methanol system; Flow rate: 12mL/min; O.D. 230 nm) from the synthesized dehydrophenylahistin of Example 1 (Fig 2) and the above exemplified tBu-dehydrophenylahistin compound produced by Route A.
[0220] The sequence of introduction of the aldehydes is a relevant to the yield and is therefore aspect of the synthesis. An analogue of dehydrophenylahistin was synthesized, as a confrol or model, wherein the dimethylallyl group was changed to the tert-butyl group with a similar steric hindrance at the 5-position of the imidazole ring.
[0221] The synthesis of this “tert-butyl (tBu)-dehydrophenylahistin” using “Route A” was as shown above: Particularly, the sequence of infroduction of the aldehyde exactly follows the dehydrophenylahistin synthesis, and exhibited a total yield of 16.5% tBu-dehydrophenylahistin. This yield was similar to that of dehydrophenylahistin (20%). Using “Route B”, where the sequence of introduction of the aldehydes is opposite that of Route “A” for the dehydrophenylahistin synthesis, only a trace amount of the desired tBu-dehydroPLH was obtained with a total yield of 0.9%, although in the introduction of first benzaldehyde 4 gave a 76% yield of the intermediate compound 17. This result indicated that it may be difficult to introduce the highly bulky imidazole-4-carboxaldehydes 15 with a substituting group having a quaternary-carbon on the adjacent 5-position at the imidazole ring into the intermediate compound 17, suggesting that the sequence for introduction of aldehydes is an important aspect for obtaining a high yield of dehydrophenylahistin or an analog of dehydrophenylahistin employing the synthesis disclosed herein:
[0222] From the HPLC analysis of the final crude products, as shown in Figure 4, a very high content of tBu-dehydrophenylahistin and small amount of by-product formations were observed in the crude sample of Route A (left). However, a relatively smaller amount of the desired tBu-dehydrophenylahistin and several other by-products were observed in the sample obtained using Route B (right).
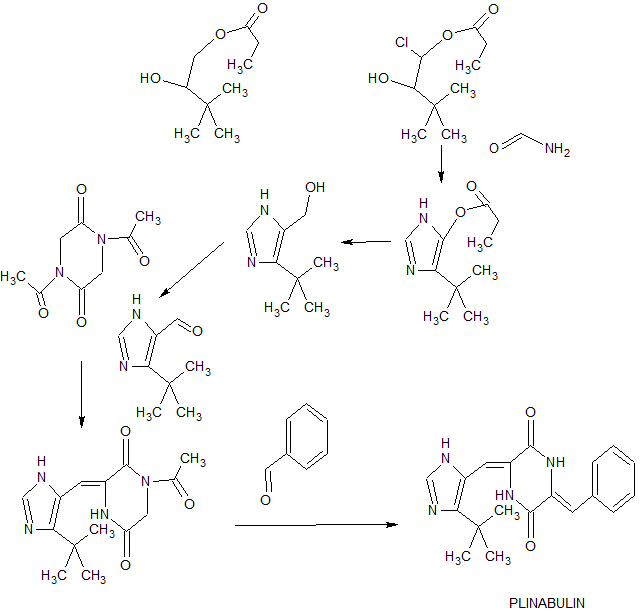
Synthesis oƒ 3-Z-Benzylidene-6-(5″-tert-butyl-1H-imidazol-4″-Z-ylmethylene)-piperazine-2,5-dione (2)

Reagents: g) SO2Cl2; h) H2NCHO, H2O; I)LiAlH4; j) MnO2; k) 1,4-diacetyl-piperazine-2,5-dione, Cs2CO3; 1) benzaldehyde, Cs2CO3
2-Chloro-4,4-dimethyl-3-oxo-pentanoic acid ethyl ester

[0280] Sulfuryl chloride (14.0 ml, 0.17 mol) was added to a cooled (0°) solution of ethyl pivaloylacetate (27.17 g, 0.16 mol) in chloroform (100 ml). The resulting mixture was allowed to warm to room temperature and was stirred for 30 min, after which it was heated under reflux for 2.5 h. After cooling to room temperature, the reaction mixture was diluted with chloroform, then washed with sodium bicarbonate, water then brine.
[0281] The organic phase was dried and evaporated to afford, as a clear oil, 2-chloro-4,4-dimethyl-3-oxo-pentanoic acid ethyl ester (33.1 g, 102%). (Durant et al., “Aminoalkylimidazoles and Process for their Production.” Patent No. GB1341375 (Great Britain, 1973)).
[0282] HPLC (214nm) tR = 8.80 (92.9%) min.
[0283] 1H NMR (400 MHz, CDCl3) δ 1.27 (s, 9H); 1.29 (t, J= 7.2 Hz, 3H); 4.27
(q, J= 7.2 Hz, 2H); 5.22 (s, 1H).
[0284] 13C NMR (100 MHz, CDCl3) δ 13.8, 26.3, 45.1, 54.5, 62.9, 165.1, 203.6.
5-tert-Butyl-3H-imidazole-4-carboxylic acid ethyl ester

[0285] A solution of 2-chloro-4,4-dimethyl-3-oxo-pentanoic acid ethyl ester (25.0 g, 0.12 mol) in formamide (47.5 ml) and water (2.5 ml) was shaken, then dispensed into 15 x 8 ml vials. All vials were sealed and then heated at 150° for 3.5 h. The vials were allowed to cool to room temperature, then water (20 ml) was added and the mixture was exhaustively extracted with chloroform. The chloroform was removed to give a concentrated formamide solution (22.2 g) which was added to a flash silica column (6 cm diameter, 12 cm height) packed in 1% MeOH/1% Et3N in chloroform. Elution of the column with 2.5 L of this mixture followed by 1 L of 2% MeOH/1% Et3N in chloroform gave, in the early fractions, a product suspected of being 5-tert-butyl-oxazole-4-carboxylic acid ethyl ester (6.3 g, 26%).
[0286] HPLC (214nm) tR = 8.77 min.
[0287] 1H NMR (400 MHz, CDCl3) δ 1.41 (t, J= 7.2 Hz, 3H); 1.43 (s, 9H); 4.40
(q, J= 7.2 Hz, 2H); 7.81 (s, 1H).
[0288] 13C NMR (100 MHz, CDCl3) δ 14.1, 28.8, 32.5, 61.3, 136.9, 149.9, 156.4,
158.3.
[0289] ESMS m/z 198.3 [M+H]+, 239.3 [M+CH4CN]+.
[0290] LC/MS tR = 7.97 (198.1 [M+H]+) min.
[0291] Recovered from later fractions was 5-tert-butyl-3H-imidazole-4-carboxylic acid ethyl ester (6.20 g, 26%). (Durant et al., “Aminoalkylimidazoles and Process for their Production.” Patent No. GB 1341375 (Great Britain, 1973)).
[0292] HPLC (214nm) tR = 5.41 (93.7%) min.
[0293] 1H NMR (400 MHz, CDCl3) δ 1.38 (t, J = 7.0 Hz, 3H); 1.47 (s, 9H); 4.36
(q, J= 7.2 Hz, 2H); 7.54 (s, 1H).
[0294] 13C NMR (100 MHz, CDCl3) δ 13 7, 28.8, 32.0, 59.8, 124.2, 133.3, 149.2,
162.6.
[0295] ESMS m/z 197.3 [M+H]+, 238.3 [M+CH4CN]+.
[0296] Further elution of the column with 1L of 5% MeOh/1% Et3N gave a compound suspected of being 5-tert-butyl-3H-imidazole-4-carboxylic acid (0.50 g, 2%).
[0297] HPLC (245nm) tR = 4.68 (83.1%) min.
[0298] 1H NMR (400 MHz, CD3OD) δ 1.36 (s, 9H); 7.69 (s, 1H).
[0299] 1H NMR (400 MHz, CDCl3) δ 1.37 (s, 9H); 7.74 (s, 1H).
[0300] 1H NMR (400 MHz, CD3SO) δ 1.28 (s, 9H); 7.68 (s, 1H).
[0301] ESMS m/z 169.2 [M+H]+, 210.4 [M+CH4CN]+.
(5-tert-Butyl-3H-imidazol-4-yl)-methanol

[0302] A solution of 5-tert-butyl-3-imidazole-4-carboxylic acid ethyl ester (3.30 g, 16.8 mmol) in THF (60 ml) was added dropwise to a suspension of lithium aluminium hydride (95% suspension, 0.89 g, 22.2 mmol) in THF (40 ml) and the mixture was stirred at room temperature for 3 h. Water was added until the evolution of gas ceased, the mixture was stirred for 10 min, then was filtered through a sintered funnel. The precipitate was washed with THF, then with methanol, the filtrate and washings were combined and evaporated. The residue was freeze-dried overnight to afford, as a white solid (5-tert-butyl- 3H-imidazol-4-yl)-methanol (2.71 g, 105%). (Durant et al., “Aminoalkylimidazoles and Process for their Production.” Patent No. GB1341375 (Great Britain, 1973)).
[0303] HPLC (240nm) tR = 3.70 (67.4%) min.
[0304] 1H NMR (400 MHz, CD3OD) δ 1 36 (s, 9H). 4 62 (s, 2H); 7.43 (s, 1H).
[0305] 13C NMR (100 MHz, CD3OD) δ 31.1, 33.0, 57.9, 131.4, 133.9, 140.8.
[0306] LC/MS tR = 3.41 (155.2 [M+H]+) min.
[0307] This material was used without further purification.
5-tert-Butyl-3H-imidazole-4-carbaldehyde

[0308] Manganese dioxide (30 g, 0.35 mol) was added to a heterogeneous solution of (5-tert-butyl-3H-imidazol-4-yl)-methanol (4.97 g, 0.03 mol) in acetone (700 ml) and the resulting mixture was stirred at room temperature for 4 h. The mixture was filtered through a pad of Celite and the pad was washed with acetone. The filfrate and washings were combined and evaporated. The residue was triturated with ether to afford, as a colorless solid, 5-tert-butyl-3H-imidazole-4-carbaldehyde (2.50 g, 51%). (Hayashi, Personal Communication (2000)).
[0309] HPLC (240nm) tR = 3.71 (89.3%) min.
[0310] 1H NMR (400 MHz, CDCl3) δ 1.48 (s, 9H); 7.67 (s, 1H); 10.06 (s, 1H).
[0311] LC/MS tR = 3.38 (153.2 [M+H]+) min.
[0312] Evaporation of the filtrate from the trituration gave additional 5-tert-butyl-3H-imidazole-4-carbaldehyde (1.88 g, 38%).
1-Acetyl-3-(5′-tert-butyl-1H-imdazol-4′-Z-ylmethylene)-piperazine-2,5-dione

[0313] To a solution of 5-tert-butyl-3H-imidazole-4-carbaldehyde (2.50 g, 164.4 mmol) in DMF (50 ml) was added 1,4-diacetyl-piperazine-2,5-dione (6.50 g, 32.8 mmol) and the solution was evacuated, then flushed with argon. The evacuation-flushing process was repeated a further two times, then cesium carbonate (5.35 g, 16.4 mmol) was added. The evacuation-flushing process was repeated a further three times, then the resultant mixture was stirred at room temperature for 5 h. The reaction mixture was partially evaporated (heat and high vacuum) until a small volume remained and the resultant solution was added dropwise to water (100 ml). The yellow precipitate was collected, then freeze-dried to afford 1-acetyl-3-(5′-tert-butyl-1Η-imidazol-4′-Z-ylmethylene)-piperazine-2,5-dione (2.24 g, 47%). (Hayashi, Personal Communication (2000)).
[0314] HPLC (214nm) tR = 5.54 (94.4%) min.
[0315] 1H NMR (400 MHz, CDCl3) δ 1.47 (s, 9H); 2.65 (s, 3H), 4.47 (s, 2H);
7.19 (s, 1H); 7.57 (s, 1H), 9.26 (s, 1H), 12.14 (s, 1H).
[0316] 13C NMR (100 MHz, CDCI3+CD3OD) δ 27.3, 30.8, 32.1, 46.5, 110.0,
123.2, 131.4, 133.2, 141.7, 160.7, 162.8, 173.0
[0317] LC/MS tR = 5.16 (291.2 [M+H]+, 581.6 [2M+H]+) min.
3-Z-Benzylidene-6-(5″-tert-butyl-lH-imidazol-4″-Z-ylmethylene)-piperazine-2,5-dione

[0318] To a solution of 1-acetyl-3-(5′-tert-butyl-1H-imidazol-4′-Z-ylmethylene)-piperazine-2,5-dione (2.43 g, 8.37 mmol) in DMF (55 ml) was added benzaldehyde (4.26 ml, 41.9 mmol) and the solution was evacuated, then flushed with nitrogen. The evacuation-
flushing process was repeated a further two times, then cesium carbonate (4.09 g, 12.6 mmol) was added. The evacuation-flushing process was repeated a further three times, then the resultant mixture was heated under the temperature gradient as shown below. After a total time of 5 h the reaction was allowed to cool to room temperature and the mixture was added to ice-cold water (400 ml). The precipitate was collected, washed with water, then freeze-dried to afford a yellow solid (2.57 g, HPLC (214nm) tR = 6.83 (83.1%) min.). This material was dissolved in chloroform (100 ml) and evaporated to azeofrope remaining water, resulting in a brown oil. This was dissolved in chloroform (20 ml) and cooled in ice. After 90 min the yellow precipitate was collected and air-dried to afford 3-Z-benzylidene-6-(5″-tert-butyl-1H-imidazol-4″-Z-ylmethylene)-piperazine-2,5-dione (1.59 g, 56%). (Hayashi, Personal Communication (2000)).
[0319] HPLC (214nm) tR = 6.38 (2.1%), 6.80 (95.2) min.
[0320] 1H NMR (400 MHz, CDCl3) δ 1.46 (s, pH). 7 01 (s, 1H, -C-C=CH); 7.03
(s, 1H, -C-C=CH); 7.30-7.50 (m, 5H, Ar); 7.60 (s, 1H); 8.09 (bs, NH); 9.51 (bs, NH); 12.40 (bs, NH).
[0321] LC/MS tR = 5.84 (337.4 [M+H]+, E isomer), 6.25 (337.4 [M+H]+, 673.4 [2M+H]+, Z isomer) min.
[0322] ESMS m/z 337.3 [M+H]+, 378.1 [M+OLGNT.
[0323] Evaporation of the chloroform solution gave additional 3-Z-benzylidene-6-(5″-tert-butyl-1H-imidazol-4″-Z-ylmethylene)-piperazine-2,5-dione (0.82 g, 29%). ΗPLC (214nm) tR = 6.82 (70.6%) min.
PAPER
Journal of Medicinal Chemistry (2012), 55(3), 1056-1071

Plinabulin (11, NPI-2358) is a potent microtubule-targeting agent derived from the natural diketopiperazine “phenylahistin” (1) with a colchicine-like tubulin depolymerization activity. Compound 11 was recently developed as VDA and is now under phase II clinical trials as an anticancer drug. To develop more potent antimicrotubule and cytotoxic derivatives based on the didehydro-DKP skeleton, we performed further modification on the tert-butyl or phenyl groups of 11, and evaluated their cytotoxic and tubulin-binding activities. In the SAR study, we developed more potent derivatives 33 with 2,5-difluorophenyl and 50 with a benzophenone in place of the phenyl group. The anti-HuVEC activity of 33 and 50 exhibited a lowest effective concentration of 2 and 1 nM for microtubule depolymerization, respectively. The values of 33 and 50 were 5 and 10 times more potent than that of CA-4, respectively. These derivatives could be a valuable second-generation derivative with both vascular disrupting and cytotoxic activities.
Synthesis and Structure–Activity Relationship Study of Antimicrotubule Agents Phenylahistin Derivatives with a Didehydropiperazine-2,5-dione Structure

PAPER
Chemistry – A European Journal (2011), 17(45), 12587-12590, S12587/1-S12587/13
Abstract

Click for improved solubility: A water-soluble prodrug of plinabulin was designed and synthesized efficiently by using click chemistry in three steps (see scheme). The product was highly water-soluble, and the parent compound could be regenerated by esterase hydrolysis.
PATENT
WO2017011399, PLINABULIN COMPOSITIONS
References
- “Assessment of Docetaxel + Plinabulin Compared to Docetaxel + Placebo in Patients With Advanced NSCLC With at Least One Measurable Lung Lesion (DUBLIN-3)”.
- Lloyd, G.K.; Muller, Ph.; Kashyap, A.; Zippelius, A.; Huang, L. (January 7–9, 2016), Plinabulin: Evidence for an Immune Mediated Mechanism of Action (Philadelphia (PA) AACR 2016 Abstract nr A07), San Diego CA
- Singh, A.V.; Bandi, M.; Raje, N.; Richardson, P.; Palladino, M.A.; Chauhan, D.; Anderson, K. (2011). “A Novel Vascular Disrupting Agent Plinabulin Triggers JNK-Mediated Apoptosis and Inhibits Angiogenesis in Multiple Myeloma Cells”. Blood. 117 (21): 5692–5700.
- Heist, R.S.; Aren, O.R.; Mita, A.C.; Polikoff, J.; Bazhenova, L.; Lloyd, G.K.; Mikrut, W.; Reich, W.; Spear, M.A.; Huang, L. (2014), Randomized Phase 2 Trial of Plinabulin (NPI-2358) Plus Docetaxel in Patients with Advanced Non-Small Lung Cancer (NSCLC) (abstr 8054)
- “Nivolumab and Plinabulin in Treating Patients With Stage IIIB-IV, Recurrent, or Metastatic Non-small Cell Lung Cancer”.
- “Nivolumab in Combination With Plinabulin in Patients With Metastatic Non-Small Cell Lung Cancer (NSCLC)”.
- Lloyd, G.K.; Du, L.; Lee, G.; Dalsing-Hernandez, J.; Kotlarczyk, K.; Gonzalez, K.; Nawrocki, S.; Carew, J.; Huang, L. (October 5–9, 2015), Activity of Plinabulin in Tumor Models with Kras Mutations (Philadelphia (PA) AACR 2015 Abstract nr. 184), Boston MA
 |
|
| Names | |
|---|---|
| IUPAC name
(3Z,6Z)-3-Benzylidene-6-{[5-(2-methyl-2-propanyl)-1H-imidazol-4-yl]methylene}-2,5-piperazinedione
|
|
| Identifiers | |
| 714272-27-2 |
|
| 3D model (Jmol) | Interactive image |
| ChemSpider | 8125252 |
| PubChem | 9949641 |
| Properties | |
| C19H20N4O2 | |
| Molar mass | 336.40 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
////////////Plinabulin, Phase 3, Clinical, 714272-27-2, NPI 2358, Nereus, (S)-(-)-phenylahistin, NPI-2350, (-)-phenylahistin, KPU-2, KPU-02, KPU-35
O=C3N\C(=C/c1ncnc1C(C)(C)C)C(=O)N/C3=C\c2ccccc2