














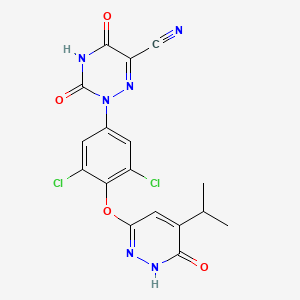
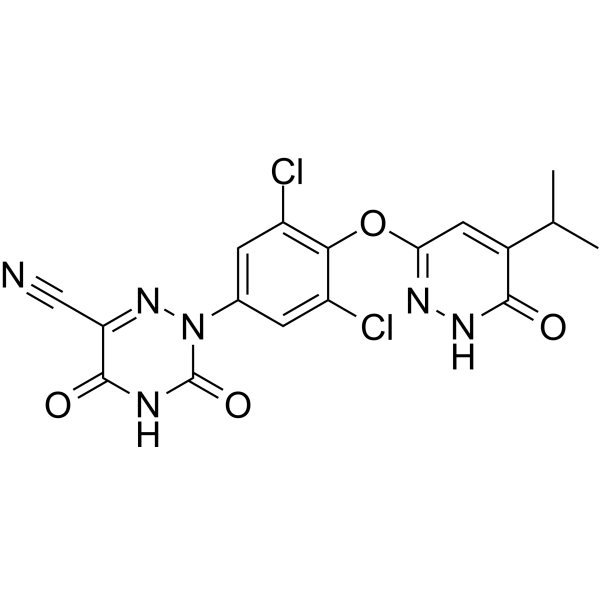

RESMETIROM
| C17H12Cl2N6O4 |
435.2 g/mol
MGL-3196
CAS 920509-32-6, Resmetirom, VIA-3196, UNII-RE0V0T1ES0
Phase III, Non-alcoholic fatty liver disease (NAFLD)
2-[3,5-dichloro-4-[(6-oxo-5-propan-2-yl-1H-pyridazin-3-yl)oxy]phenyl]-3,5-dioxo-1,2,4-triazine-6-carbonitrile
2-(3,5-DICHLORO-4-((5-ISOPROPYL-6-OXO-1,6-DIHYDROPYRIDAZIN-3-YL)OXY)PHENYL)-3,5-DIOXO-2,3,4,5-TETRAHYDRO-(1,2,4)TRIAZINE-6-CARBONITRILE
1,2,4-TRIAZINE-6-CARBONITRILE, 2-(3,5-DICHLORO-4-((1,6-DIHYDRO-5-(1-METHYLETHYL)-6-OXO-3-PYRIDAZINYL)OXY)PHENYL)-2,3,4,5-TETRAHYDRO-3,5-DIOXO-
Madrigal Pharmaceuticals , following the merger between Synta and Madrigal Pharmaceuticals (pre-merger) (following the acquisition of VIA Pharmaceuticals ‘ assets (originally under license from Roche )), is developing resmetirom (MGL-3196, VIA-3196), the lead from oral capsule formulation thyroid hormone receptor (THR) beta agonists, cholesterol and triglyceride modulators, for the use in the treatment of metabolic disorders including hypercholesterolemia and other dyslipidemias, and non-alcoholic steatohepatitis.
MGL-3196 is a first-in-class, orally administered, small-molecule, liver-directed, THR β-selective agonist. Preclinical, toxicology and Phase 1 clinical data suggest MGL-3196 has an attractive, differentiated profile as a potential treatment for non-alcoholic steatohepatitis (NASH) and dyslipidemias. THR-β selectivity also enhances the safety profile of MGL-3196, compared to non-selective agents. MGL-3196 has shown no suppression of the central thyroid axis, no THR-α effects on heart rate or bone, and no elevation of liver enzymes. These characteristics make MGL-3196 among the most promising molecules in development in this therapeutic area. MGL-3196 is in a Phase 2 clinical trial for the treatment of non-alcoholic steatohepatitis (NASH).
PATENT
WO-2020010068
Novel crystalline salt of resmetirom as thyroid hormone receptor agonists useful for treating obesity, hyperlipidemia, hypercholesterolemia and diabetes. Appears to be the first filing from the assignee and the inventors on this compound,
Thyroid hormones are critical for normal growth and development and for maintaining metabolic homeostasis (Paul M. Yen, Physiological reviews, Vol. 81(3): pp. 1097-1126 (2001)). Circulating levels of thyroid hormones are tightly regulated by feedback mechanisms in the hypothalamus/pituitary/thyroid (HPT) axis. Thyroid dysfunction leading to hypothyroidism or hyperthyroidism clearly demonstrates that thyroid hormones exert profound effects on cardiac function, body weight, metabolism, metabolic rate, body temperature, cholesterol, bone, muscle and behavior.
[0005] The biological activity of thyroid hormones is mediated by thyroid hormone receptors (TRs or THRs) (M. A. Lazar, Endocrine Reviews, Vol. 14: pp. 348-399 (1993)). TRs belong to the superfamily known as nuclear receptors. TRs form heterodimers with the retinoid receptor that act as ligand-inducible transcription factors. TRs have a ligand binding domain, a DNA binding domain, and an amino terminal domain, and regulate gene expression through interactions with DNA response elements and with various nuclear co-activators and co repressors. The thyroid hormone receptors are derived from two separate genes, a and b. These distinct gene products produce multiple forms of their respective receptors through differential RNA processing. The major thyroid receptor isoforms are aΐ, a2, bΐ, and b2. Thyroid hormone receptors aΐ, bΐ, and b2 bind thyroid hormone. It has been shown that the thyroid hormone receptor subtypes can differ in their contribution to particular biological responses. Recent studies suggest that TIIb 1 plays an important role in regulating TRH (thyrotropin releasing hormone) and on regulating thyroid hormone actions in the liver. T11b2 plays an important role in the regulation of TSH (thyroid stimulating hormone) (Abel et. al, J. Clin. Invest., Vol 104: pp. 291-300 (1999)). TIIb 1 plays an important role in regulating heart rate (B. Gloss et. al. Endocrinology, Vol. 142: pp. 544-550 (2001); C. Johansson et. al, Am. J. Physiol., Vol. 275: pp. R640-R646 (1998)).
[0006] Efforts have been made to synthesize thyroid hormone analogs which exhibit increased thyroid hormone receptor beta selectivity and/or tissue selective action. Such thyroid hormone mimetics may yield desirable reductions in body weight, lipids, cholesterol, and lipoproteins, with reduced impact on cardiovascular function or normal function of the hypothalamus/pituitary/thyroid axis (see, e.g., Joharapurkar et al, J. Med. Chem, 2012, 55 (12), pp 5649-5675). The development of thyroid hormone analogs which avoid the undesirable effects of hyperthyroidism and hypothyroidism while maintaining the beneficial effects of thyroid hormones would open new avenues of treatment for patients with metabolic disease such as obesity, hyperlipidemia, hypercholesterolemia, diabetes and other disorders and diseases such as liver steatosis and NASH, atherosclerosis, cardiovascular diseases, hypothyroidism, thyroid cancer, thyroid diseases, a resistance to thyroid hormone (RTH) syndrome, and related disorders and diseases.
PATENT
WO2018075650
In one embodiment, the metabolite of Compound A comprises a compound
having the following structure:
(“Ml”).
PATENT
WO 2007009913
PATENT
WO 2014043706
https://patents.google.com/patent/WO2014043706A1/en
Example 3: Preparation of (Z)-ethyl (2-cyano-2-(2-(3,5-dichloro-4-((5-isopropyl-6- oxo- l,6-dihydropyridazin-3-yl)oxy)phenyl)hydrazono)acetyl)carbamate (Int. 8)
A 2 L, three-neck, round-bottom flask equipped with overhead stirring, a thermocouple, N2 inlet/outlet was charged with Int. 7 (75.0 g, 0.239 mol, 1 wt), acetic acid (600 mL, 8 vol), water (150 mL, 2 vol), and concentrated HC1 (71.3 mL, 0.95 vol). The resulting thin slurry was cooled to 6 °C and a solution of NaN02 (16.8 g, 0.243 mol, 1.02 equiv) in water (37.5 mL, 0.5 vol) was added over a period of 10 min while maintaining a batch temperature below 10 °C. After an additional 10 min of agitation between 5-10 °C, HPLC analysis showed complete conversion of Int. 7 to the diazonium intermediate. A solution of NaOAc (54.5 g, 0.664 mol, 2.78 equiv) in water (225 mL, 3 vol) was added over a period of 6 min while maintaining a batch temperature below 10 °C. N-cyanoacetylurethane (37.9 g, 0.243 mol, 1.02 equiv) was immediately added, the cooling was removed, and the batch naturally warmed to 8 °C over 35 min. HPLC analysis showed complete consumption of the diazonium intermediate and the reaction was deemed complete. The batch warmed naturally to 21 °C and was filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with water (375 mL, 5 vol) twice. The collected orange solid was dried in a 35 °C vacuum oven for 64 h to provide crude Int. 8 (104.8 g, 91%).
A I L, three-neck, round-bottom flask equipped with overhead stirring, a
thermocouple, and N2 inlet/outlet was charged with crude Int. 8 (104.4 g, 1 wt) and acetic acid (522 mL, 5 vol). The resulting slurry was heated to 50 °C and held at that temperature for 1.5 h. The batch cooled naturally to 25 °C over 2 h and was filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with water (522 mL, 5 vol) and the cake conditioned under vacuum for 1.75 h. The light orange solid was dried to constant weight in a 40 °C vacuum oven to provide 89.9 g (78% from Int. 7) of the desired product. 1H NMR (DMSO) was consistent with the assigned structure.
Example 4: Preparation of 2-(3,5-dichloro-4-((5-isopropyl-6-oxo-l,6- dihydropyridazin-3-yl)oxy)phenyl)-3,5-dioxo-2,3,4,5-tetrahydro-l,2,4-triazine-6-carbonitrile (Compound A)
A 2 L, three-neck, round-bottom flask equipped with overhead stirring, a
thermocouple, N2 inlet/outlet, and reflux condenser was charged with Int. 8 (89.3 g, 0.185 mol, 1 wt), DMAC (446 mL, 5 vol), and KOAc (20.0 g, 0.204 mol, 1.1 equiv). The mixture was heated to 120 °C and held at that temperature for 2 h. HPLC analysis showed complete conversion to Compound A. The batch temperature was adjusted to 18 °C over 1 h, and acetic acid (22.3 mL, 0.25 vol) was added. The batch temperature was adjusted to 8 °C, and water (714 mL, 8 vol) was added over 1 h; an orange slurry formed. The batch was filtered through Sharkskin filter paper and the cake was allowed to condition overnight under N2 without vacuum for convenience. A premixed solution of 1 : 1 acetone/water (445 mL, 5 vol) was charged to the flask and added to the cake as a rinse with vacuum applied. After 2 h of conditioning the cake under vacuum, it was transferred to a clean 1 L, three-neck, round- bottom flask equipped with overhead stirring, a thermocouple, and N2inlet/outlet. Ethanol (357 mL, 4 vol) and acetone (357 mL, 4 vol) were charged and the resulting slurry was heated to 60 °C; dissolution occurred. Water (890 mL, 10 vol) was added over a period of 90 min while maintaining a batch temperature between 55-60 °C. The resulting slurry was allowed to cool to 25 °C and filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with a solution of 1:1 EtOH/water (446 mL, 5 vol). The cake was conditioned overnight under N2 without vacuum for convenience. The cracks in the cake were smoothed and vacuum applied. The cake was washed with water (179 mL, 2 vol) and dried in a 45 °C vacuum oven to a constant weight of 70.5 g (87%, crude Compound A). HPLC analysis showed a purity of 94.8%.
A 500 mL, three-neck, round-bottom flask equipped with overhead stirring, a thermocouple, N2 inlet/outlet, and reflux condenser was charged with crude Compound A (70.0 g) and MIBK (350 mL, 5 vol). The orange slurry was heated to 50 °C and held at that temperature for 2 h. The batch cooled naturally to 23 °C and was filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with MIBK (35 mL, 0.5 vol) twice. The collected solids were dried in a 45 °C vacuum oven to a constant weight of 58.5 g (84%). This solid was charged to a 500 mL, three-neck, round-bottom flask equipped with overhead stirring, a thermocouple, N2 inlet/outlet, and reflux condenser. Ethanol (290 mL, 5 vol) was added and the slurry was heated to reflux. After 3.5 h at reflux, XRPD showed the solid was consistent with Form I, and heating was removed. Upon reaching 25 °C, the batch was filtered through filter paper, and the reactor and cake were washed sequentially with EtOH (174 mL, 3 vol). The tan solid Compound A was dried in a 40 °C vacuum oven to a constant weight of 50.4 g (87%, 64% from Int. 8). HPLC analysis showed a purity of 99.1%. 1H NMR (DMSO) was consistent with the assigned structure.
Example 5: Scaled up preparation of 2-(3,5-dichloro-4-((5-isopropyl-6-oxo-l,6- dihydropyridazin-3-yl)oxy)phenyl)-3,5-dioxo-2,3,4,5-tetrahydro-l,2,4-triazine-6-carbonitrile (Compound A)
A larger scale batch of Compound A was synthesized according to the scheme below. The conditions in the scheme below are similar to those described in Examples 1-4 above.

6A

Compound A
Synthesis of 4: A 50 L jacketed glass vessel (purged with N2) was charged with 3,6- dichloropyridazine (2.00 kg), 4-amino-2,6-dichlorophenol (2.44 kg) and N,N- dimethylacetamide (10.0 L). The batch was vacuum (26 inHg) / nitrogen (1 PSIG) purged 3 times. Cesium carbonate (5.03 kg) was added and the batch temperature was adjusted from 22.3 °C to 65.0 °C over 3.5 hours. The batch was held at 65.0 °C for 20 hours. At this point,
NMR analysis indicated 3.34% 3.6-dichloropyridazine relative to 2. The batch temperature was adjusted to 21.5 °C and ethyl acetate (4.00 L) was added to the batch. The batch was agitated for 10 minutes and then filtered through a 18″ Nutsche filter equipped with polypropylene filter cloth. The filtration took 15 minutes. Ethyl acetate (5.34 L) was charged to the vessel and transferred to the filter as a rinse. The batch was then manually re- suspended in the filter before re-applying vacuum. This process was repeated 2 more times and the filter cake was conditioned for 10 minutes. The filtrate was charged to a 100-L vessel that contained (16.0 L) of a previously prepared 15% sodium chloride in H20. The batch was agitated for 5 minutes and then allowed to separate for 35 minutes. The interface was not visible, so the calculated 23 L of the lower aqueous phase was removed. 16.0 L of 15% Sodium chloride in H20 was added to the batch. The batch was agitated for 6 minutes and then allowed to separate for 7 minutes. The interface was visible at -19 L and the lower aqueous phase was removed. 17.0 L of 15% Sodium chloride in H20 was added to the batch. The batch was agitated for 7 minutes and then allowed to separate for 11 minutes. The lower aqueous phase was removed. The vessel was set up for vacuum distillation and the batch was concentrated from 17.0 L to 8.0 L over 2 hours 20 minutes with the batch temperature kept around 21 °C. Benzoic anhydride (3.19 kg) and acetic acid (18.0 L) were charged to the vessel. The vessel was set up for vacuum distillation and the batch was concentrated from 28.0 L to 12.0 L over 2 days (overnight hold at 20 °C) with the batch temperature kept between 20 and 55 °C. At this point, JH NMR analysis indicated a mol ratio of acetic acid to ethyl acetate of 1.0:0.015. Acetic acid (4.0 L) was charged to the batch and the batch was distilled to 12 L. JH NMR analysis indicated a mol ratio of acetic acid to ethyl acetate of 1.0:0.0036. Acetic acid (20.0 L) was charged to the batch and the batch temperature was adjusted to 70.0 °C. The batch was sampled for HPLC analysis and 2 was 0.16%. Sodium acetate (2,20 kg) was added to the batch and the batch temperature was adjusted from 72.4 °C to 110.0 °C. After 18.5 hours, HPLC analysis indicated no Int. B detected. The batch temperature was adjusted from 111.3 to 74.7 °C and DI water (30.0 L) was added to the batch over 2 hours. The batch temperature was adjusted to 20 .5 °C and then filtered using a 24″ Haselloy Nutsche filter equipped with polypropylene filter cloth. A previously prepared solution of 1:1 acetic acid in DI H20 (10.0 L) was charged to the vessel and agitated for 5 minutes. The wash was transferred to the filter and the batch was then manually re- suspended in the filter before re-applying vacuum. DI H20 (10.0 L) was charged to the vessel and then transferred to the filter. The batch was manually re-suspended in the filter before re-applying vacuum. DI H20 (10.0 L) was charged directly to the filter and the batch was then manually re-suspended in the filter before re-applying vacuum. The filter cake was allowed to condition for 18 hours to give 14.4 kg of 4. HPLC analysis indicated a purity of 93.7%. This wet cake was carried forward into the purification. A 100 L jacketed glass vessel (purged with N2) was charged with crude 4 (wet cake 14.42 kg), acetic acid (48.8 L) and the agitator was started. DI H20 (1.74 L) was charged. The batch (a slurry) temperature was adjusted from 18.1 to 100.1 °C over 4.25 hours. The batch was held at 100.1 to 106.1 °C for 1 hour and then adjusted to 73.1 °C. DI H20 (28.0 L) was added to the batch over 1 hour keeping the batch temperature between 73.1 and 70.3 °C. The batch temperature was adjusted further from 70.3 °C to 25.0 °C overnight. The batch was filtered using a 24″ Hastelloy Nutsche filter equipped with polypropylene filter cloth. The filtration took 13 minutes. A solution of DI H20 (9.00 L) and acetic acid (11.0 L) was prepared and added to the 100 L vessel. The mixture was agitated for 5 minutes and then transferred to the filter cake. DI H20 (20.0 L) was charged to the vessel, agitated for 6 minutes and then transferred to the filter cake. DI H20 (20.0 L) was charged to the vessel, agitated for 9 minutes and then transferred to the filter cake. The batch was allowed to condition for 3 days and then transferred to drying trays for vacuum oven drying. After 3 days at 50 °C and 28’7Hg, the batch gave a 74% yield (3.7 kg) of4 as an off-white solid. The JH NMR spectrum was consistent with the assigned structure, HPLC analysis indicated a purity of 98.87% and KF analysis indicated 0.14% H20. Synthesis of Int. 7: A 100-L jacketed glass vessel (purged with N2) was charged with tetrahydrofuran (44.4 L). The agitator was started (125 RPM) and 4 (3.67 kg) was charged followed by lithium chloride (1.26 kg). The batch temperature was observed to be 26.7 ° C and was an amber solution. Isopropenylmagnesium bromide 1.64 molar solution in 2-methyl THF (21.29 kg) was added over 2 ½ hours keeping the batch between 24.3 and 33.6 °C. The batch was agitated at 24.5 °C for 17 hours at which point HPLC analysis indicated 9% 4. A 2nd 100-L jacketed glass vessel (purged with N2) was charged with 3N hydrogen chloride (18.3 L). The batch was transferred to the vessel containing the 3N HC1 over 25 minutes keeping the batch temperature between 20 and 46 °C. A bi-phasic solution was observed. The quenched batch was transferred back to the 1st 100-L vessel to quench the small amount of residue left behind. THF (2.00 L) was used as a rinse. The batch temperature was observed to be 40.9 ° C and was agitated at 318 RPM for 45 minutes. The batch temperature was adjusted to 21.8 ° C and the layers were allowed to separate. The separation took 10 minutes. The lower aqueous phase was removed (-26.0 L). A solution of sodium chloride (1.56 kg) in DI water (14.0 L) was prepared and added to the batch. This was agitated at 318 RPM for 10 minutes and agitator was stopped. The separation took 3 minutes. The lower aqueous phase was removed (-16.0 L). The batch was vacuum distilled from 58.0 L to 18.4 L using ~24’7Hg and a jacket temperature of 50 to 55 °C. A solution of potassium hydroxide (2.30 kg) in DI water (20.7 L) was prepared in a 72-L round bottom flask. The vessel was set up for atmospheric distillation using 2 distillation heads and the batch was transferred to the 72-L vessel. THF (0.75 L) was used as a rinse. The batch volume was -41.0 L, the temperature was adjusted to 64.1 °C and distillation started with the aid of a N2 sweep. Heating was continued to drive the batch temperature to 85.4 °C while distilling at which point the 72-L vessel was set up for reflux (batch volume was about 28.0 L at the end of the distillation). The batch was held at 85 °C for 13 hours at which point HPLC analysis indicated 0.3% compound 6A. Heating was stopped and the batch was transferred to a 100-L jacketed glass vessel. Solids were observed. The batch temperature was adjusted from 70.6 °C to 56.7 °C. A previously prepared solution of sodium hydrogen carbonate (2.82 kg) in DI water (35.0 L) was added over 80 minutes keeping the batch temperature between 56.7 and 46.7 °C. The batch pH at the end of the addition was 9.8. The batch was held at
46.7 to 49.0 °C for 40 minutes and then cooled to 25.0 °C. The batch was filtered using a 18″ stainless steel Nutsche filter. DI water (18.4 L) was charged to the vessel and transferred to the filter. The filter cake was manually re-suspended in the filter and then the liquors were removed. This process was repeated once more and the filter cake was 3″ thick. The filter cake was conditioned on the filter for 3 days, was transferred to drying trays and dried in a vacuum oven at 45 °C to provide 2.93 kg Int. 7 (95% yield) with an HPLC purity of 87.6%.
Synthesis of Int. 8: A 100 L jacketed glass vessel (purged with N2 and plumbed to a caustic scrubber) was charged with acidic acid (13.0 L). Int. 7 (2.85 kg) was charged to the vessel and the agitator was started. N-Cyanoacetylurethane (1.56 kg) and DI water (5.70 L) were charged to the vessel. The batch temperature was adjusted from 17.0 °C to 5.5 °C and a thin slurry was observed. At this point 37% hydrogen chloride (2.70 L) was added over 10 minutes keeping the batch temperature between 4.8 °C and 8.8 °C. A previously prepared solution of sodium nitrite (638 g) in DI water (1.42 L) was added over 26 minutes keeping the batch temperature between 5.8 °C and 8.7 °C. A brown gas was observed in the vessel head space during the addition. HPLC analysis indicated no Int. 7 detected. At this point a previously prepared solution of sodium acetate (2.07 kg) in DI water (8.50 L) was added over 47 minutes keeping the batch temperature between 5.5 °C and 9.5 °C. After the addition, a thin layer of orange residue was observed on the vessel wall just above the level of the batch. The batch temperature was adjusted from 9.4 °C to 24.5 °C and held at 25 °C (+ 5 °C) for 12 hours. The batch was filtered using a 24″ Hastelloy Nutsche filter equipped with tight-weave polypropylene filter cloth. The filtration took 30 minutes. The vessel was rinsed with 14.3 L of a 1 : 1 acidic acid / DI water. The orange residue on the reactor washed away with the rinse. The rinse was transferred to the filter where the batch was manually re-suspended. Vacuum was re-applied to remove the wash. A 2nd 1 : 1 acidic acid / DI water wash was performed as above and the batch was conditioned on the filter for 26 hours. HPLC analysis of the wet filter cake indicated purity was 90.4%. The batch was dried to a constant weight of 3.97 kg (91% yield) in a vacuum oven at 45 °C and 287Hg. Preparation of Compound A DMAC Solvate
A 100 L, jacketed, glass vessel purged with N2 was charged with Int. 8 (3.90 kg) and potassium acetate (875 g). N,N-dimethylacetamide (DMAC, 18.3 L) was charged to the vessel and the agitator was started. The batch temperature was adjusted to 115 °C over 2 h. After 2 h at 115 °C, the batch was sampled and HPLC analysis indicated 0.27% Int. 8 remained. The batch temperature was adjusted to 25.0 °C overnight. Acetic acid (975 mL) was added to the batch and the batch was agitated further for 3 h. The batch was transferred to a carboy and the vessel was rinsed clean with 800 mL of DMAC. The batch was transferred back to the 100 L vessel using vacuum through a 10 μιη in-line filter and a DMAC rinse (1.15 L) was used. The filtration was fast at the beginning but slow at the end, plugging up the filter. The batch temperature was adjusted to 11.1 °C and DI water (35.1 L) was added over 2 h 20 min, keeping the batch temperature between 5-15 °C. The batch was held for 1 h and filtered, using an 18″ Nutsche filter equipped with tight-weave
polypropylene cloth. The filtration took 15 h. A 1: 1 ethanol/DI water wash (19.5 L) was charged to the vessel, cooled to 10 °C, and transferred to the filter cake. The cake was allowed to condition under N2 and vacuum for 8 h and transferred to drying trays. The batch was dried in a vacuum oven at 45 °C and 28’7Hg to give 89% yield (3.77 kg) of Compound A DMAC solvate as an orange/tan solid. The 1H NMR spectrum was consistent with the assigned structure and Karl Fischer analysis indicated 0.49% H20. XRPD indicated the expected form, i.e., Compound A DMAC solvate. Thermogravimetric analysis (TGA) indicated 16% weight loss. HPLC analysis indicated a purity of 93.67%.
Preparation of Crude Compound A
A 100 L, jacketed, glass vessel purged with N2 was charged with Compound A
DMAC solvate (3.75 kg) and ethanol (15.0 L). The agitator was started and acetone (15.0 L) was added. The batch temperature was adjusted from 10.6 °C to 60.0 °C over 1 h. At this point, the batch was in solution. DI water was added to the batch over 1.5 h, keeping the batch temperature at 60 + 5 °C. The batch was held at 60 + 5 °C for 1 h and cooled to 23.5 °C. An 18″ Nutsche filter equipped with tight-weave (0.67 CFM) polypropylene cloth was set up and the batch was filtered. The filtration took 15 h. A 1: 1 ethanol/DI water wash (19.5 L) was charged to the vessel and transferred to the filter cake. The cake was allowed to condition under N2 and vacuum for 8 h and transferred to drying trays. The batch was dried in a vacuum oven at 45 °C and 28’7Hg for five days to give a 94% yield (2.90 kg) of Compound A as a powdery tan solid. The NMR spectrum is consistent with the assigned structure and Karl Fischer analysis indicated 6.6% H20. XRPD indicated the expected form of dihydrate. TGA indicated 6.7% weight loss. HPLC analysis indicated a purity of 96.4% (AUC).
Purification of Crude Compound A
A 50 L, jacketed, glass vessel purged with N2 was charged with Compound A crude
(2.90 kg) and methyl isobutyl ketone (14.5 L). The agitator was started and the batch temperature was adjusted from 20.2 °C to 50.4 °C over 1.5 h. The batch was held at 50 °C (+ 5 °C) for 1 h and cooled to 20-25 °C. The batch was held at 20-25 °C for 2.5 h. An 18″ Nutsche filter equipped with tight- weave (0.67 CFM) polypropylene cloth was set up and the batch was filtered. The filtration took 20 min. Methyl isobutyl ketone (MIBK, 1.45 L) was charged to the vessel and transferred to the filter cake. The cake was manually resuspended and the liquors were pulled through with vacuum. Methyl isobutyl ketone (2.90 L) was charged to the filter cake and the cake was manually resuspended. The liquors were pulled through with vacuum and the cake was conditioned with vacuum and nitrogen for 15 h. The filter cake dried into a tan, hard 18″ x 1 ½” disc. This was manually broken up and run through coffee grinders to give a 76% yield (2.72 kg) of MGL-3196 MIBK solvate as a tan, powdery solid. No oven drying was necessary. The NMR spectrum was consistent with the assigned structure and Karl Fischer analysis indicated <0.1 % H20. XRPD indicated the expected form MIBK solvate. TGA indicated 17.3% weight loss. HPLC analysis indicated a purity of 98.5%.
Example 6: Conversion of Compound A to Form I
Purified Compound A (4802 g) as a 1:1 MIBK solvate which was obtained from Int. 8 as described in Example 5 above was added into a jacketed, 100 L reactor along with 24 liters of ethanol. The resulting slurry was heated to 80 + 5 °C (reflux) over 1 h 25 min; the mixture was stirred at that temperature for 4 h 25 min. Analysis of the filtered solids at 2 h 55 min indicated that the form conversion was complete, with the XRPD spectra conforming to Form I. The mixture was cooled to 20 + 5 °C over 45 min and stirred at that temperature for 15 min. The slurry was filtered and the filter cake was washed twice with prefiltered ethanol (2 x 4.8 L). The wet cake (4.28 kg) was dried under vacuum at 40 + 5 °C for 118 h to afford 3390 g of Compound A form I.
PAPER
Journal of Medicinal Chemistry (2014), 57(10), 3912-3923
https://pubs.acs.org/doi/abs/10.1021/jm4019299
The beneficial effects of thyroid hormone (TH) on lipid levels are primarily due to its action at the thyroid hormone receptor β (THR-β) in the liver, while adverse effects, including cardiac effects, are mediated by thyroid hormone receptor α (THR-α). A pyridazinone series has been identified that is significantly more THR-β selective than earlier analogues. Optimization of this series by the addition of a cyanoazauracil substituent improved both the potency and selectivity and led to MGL-3196 (53), which is 28-fold selective for THR-β over THR-α in a functional assay. Compound 53 showed outstanding safety in a rat heart model and was efficacious in a preclinical model at doses that showed no impact on the central thyroid axis. In reported studies in healthy volunteers, 53 exhibited an excellent safety profile and decreased LDL cholesterol (LDL-C) and triglycerides (TG) at once daily oral doses of 50 mg or higher given for 2 weeks.

//////////////RESMETIROM , MGL-3196, VIA-3196, UNII-RE0V0T1ES0, Phase III
CC(C)C1=CC(=NNC1=O)OC2=C(C=C(C=C2Cl)N3C(=O)NC(=O)C(=N3)C#N)Cl