














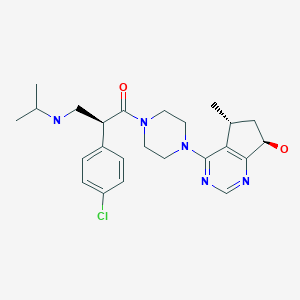
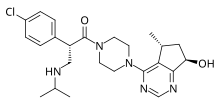
Ipatasertib
GDC-0068 , RG7440
CAS 1001264-89-6, C24H32ClN5O2, 457.9962
アイパタセルチブ;
イパタセルチブ;
Antineoplastic, AKT serine/threonine kinase inhibitor
2(S)-(4-Chlorophenyl)-1-[4-[7(R)-hydroxy-5(R)-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl]piperazin-1-yl]-3-(isopropylamino)propan-1-one
(2S)-2-(4-Chlorophenyl)-1-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta(d)pyrimidin-4-yl)piperazin-1-yl(-3-((propan-2-yl)amino)propan-1-one
1-Propanone, 2-(4-chlorophenyl)-1-(4-((5R,7R)-6,7-dihydro-7-hydroxy-5-methyl-5H-cyclopentapyrimidin-4-yl)-1-piperazinyl)-3-((1-methylethyl)amino)-, (2S)-
Ipatasertib dihydrochloride
1396257-94-5
Ipatasertib (RG7440) is an experimental cancer drug in development by Roche. It is a small molecule inhibitor of Akt. It was discovered by Array Biopharma and is currently in phase II trials for treatment of breast cancer.[1]
In vitro, ipatasertib showed activity against all three isoforms of Akt.[2]
Ipatasertib is an orally-available protein kinase B (PKB/Akt) inhibitor in phase III clinical development at Genentech for the treatment of metastatic castration-resistant prostate cancer in combination with abiraterone and prednisone.
In 2014, orphan drug designation was assigned in the U.S. for the treatment of gastric cancer including cancer of the gastro-esophageal junction.
Ipatasertib. An orally bioavailable inhibitor of the serine/threonine protein kinase Akt (protein kinase B) with potential antineoplastic activity. Ipatasertib binds to and inhibits the activity of Akt in a non-ATP-competitive manner, which may result in the inhibition of the PI3K/Akt signaling pathway and tumor cell proliferation and the induction of tumor cell apoptosis. Activation of the PI3K/Akt signaling pathway is frequently associated with tumorigenesis and dysregulated PI3K/Akt signaling may contribute to tumor resistance to a variety of antineoplastic agents. Check for active clinical trials using this agent.
PROBLEM
It has been found that ipatasertib exhibits a very high solubility (>1 g/g water; >2 g/g water/ethanol 1:1) and a very high hygroscopicity (˜6% at 50% RH, >35% at 95% RH). Whereas poor solubility is often a limiting factor in the development of galenical formulations of other API’s (active pharmaceutical ingredient), a high solubility can equally be problematic for the process performance. Due to this very high intrinsic hygroscopicity of the API, ipatasertib drug substance tends to auto-dissolve to a honey-like viscous liquid at increased humidity. Such high solubility and hygroscopicity may pose serious problems for processing as well as for stability and shelf-life of the final product. Therefore, conventional pharmaceutical compositions comprising ipatasertib and processes for the manufacture of pharmaceutical compositions comprising wetting (e.g. wet granulation) are difficult due to the high solubility and high hygroscopicity of the API.
SYN

Bromination of (+)-(R)-pulegone (I) with Br2 in the presence of NaHCO3 in Et2O, followed by ring contraction via Favorskii rearrangement with NaOEt in EtOH, and treatment with semicarbazide hydrochloride and NaOAc in refluxing EtOH/H2O gives rise to cyclopentanecarboxylate (II) (1). Subsequent ozonolysis of olefin (II) by means of O3 in EtOAc at -78 °C, and reductive treatment with Zn in AcOH provides beta-ketoester (III). Reaction of ketoester (III) with ammonium acetate (IVa) in MeOH/CH2Cl2 yields enamine (V), which upon cyclization with ammonium formate (IVb) and formamide (VI) at 150 °C provides cyclopentapyrimidinol (VII). Chlorination of pyrimidinol (VII) using POCl3 in refluxing CH2Cl2 results in 4-chloro-5(R)-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidine (VIII), which is condensed with N-Boc-piperazine (IX) in the presence of DIEA in refluxing BuOH to produce piperazinyl cyclopentapyrimidine (X). Oxidation of compound (X) using mCPBA and NaHCO3 in CHCl3 furnishes N-oxide (XI). Subsequent rearrangement of N-oxide (XI) using Ac2O in CH2Cl2 at 100 °C yields acetate (XII). This compound (XII) is hydrolyzed with LiOH in H2O/THF to give alcohol (XIII), which upon Swern oxidation with (COCl)2, DMSO and Et3N in CH2Cl2 at -78 °C affords ketone (XIV) (1-6). Asymmetric transfer hydrogenation of ketone (XIV) in the presence of RuCl[(R,R)-TsDPEN(p-cymene)], HCOOH and Et3N in CH2Cl2, followed by protection with PNBCl in the presence of Et3N in CH2Cl2, and hydrolysis with LiOH in H2O/THF gives rise to alcohol (XV) (1-6). Also, intermediate (XV) can be produced by enzymatic reduction of ketone (XI) using KRED-101 in the presence of GDH, NADP, KOH and PEG-400, KRED-X1.1-P1F01 in the presence of glucose and NAD in DMSO/i-PrOH or KRED-X1.1-P1B06, KRED-X1.1-P1F01 or KRED-X1.1-P1H10 in the presence of NADP in DMSO/i-PrOH or i-PrOH (11,12). In an alternative method, asymmetric transfer hydrogenation of ketone (XIV) in the presence of RuCl[(R,R)-MsDPEN(p-cymene)], HCOOH and Et3N in CH2Cl2, followed by O-protection of the resultant cis/trans mixture of alcohols with PNBCl and Et3N or protection with pivaloyl chloride in the presence of DIEA in CH2Cl2, followed by separation of the resulting cis/trans mixture of esters by means of HPLC. Hydrolysis of trans ester with LiOH in THF yields alcohol (XV) (11). N-Deprotection of piperazine derivative (XV) by means of HCl in CH2Cl2, i-PrOH or toluene at 62 °C provides amine dihydrochloride (XVI) (1-7,11,12), which is then coupled with aminoacid derivative (XVIIa) (1-7,11) or its sodium salt (XVIIb) (12,13) in the presence of DIEA and HBTU in CH2Cl2 or NMM and T3P in i-PrOH or toluene to produce amide (XVIII) (1-7,11-13). Finally, Boc-deprotection of precursor (XVIII) by means of HCl in MeOH/Et2O, PrOH, i-PrOH or toluene at 57 °C furnishes the target GDC-0068

Synthesis of intermediate (XVII): Condensation of methyl (4-chlorophenyl)acetate (XIX) with formaldehyde (XX) in the presence of NaOMe in DMSO gives beta-hydroxyester (XXI). Subsequent dehydration of alcohol (XXI) using MsCl and Et3N in CH2Cl2 provides arylacrylate (XXII), which upon conjugate addition with isopropylamine (XXIII) in the presence of Boc2O in THF yields N-Boc beta-aminoester (XXIV). Basic hydrolysis of ester (XXIV) using KOSiMe3 in THF generates the potassium carboxylate (XXV), which upon condensation with 4(R)-benzyl-2-oxazolidinone (XXVI) via activation with pivaloyl chloride and BuLi in THF at -78 °C affords the N-acyl oxazolidinone (XXVII) (2-6). Finally, removal of the chiral auxiliary group of (XXVII) using LiOH and H2O2 in THF/H2O furnishes the key intermediate (XVII) (1-6,11). Alternative synthesis of intermediate (XXVII): Protection of isopropylamine (XXIII) with Boc2O in toluene affords tert-butyl isopropylcarbamate (XXVIII), which upon N-alkylation with bromomethyl methyl ether (XXIX) in the presence of NaHMDS in 2-MeTHF gives tert-butyl isopropyl(methoxymethyl)carbamate (XXX) (11). Condensation of 4(R)-benzyl-2-oxazolidinone (XXVI) with 2-(4-chlorophenyl)acetyl chloride (XXXIIa) using BuLi in THF at -50 °C (1) or with 2-(4-chlorophenyl)acetic acid (XXXIIb) via activation with pivaloyl chloride and Et3N in refluxing toluene (11) affords N-acyl oxazolidinone(XXXI). After conversion of intermediate (XXXI) to its titanium enolate with TiCl4 and DIEA in CH2Cl2 at -50 °C, diastereoselective Mannich reaction with formaldehyde hemiaminal (XXX) affords adduct (XXVII)
PAPER
Synthesis of Akt inhibitor ipatasertib. Part 2. Total synthesis and first kilogram scale-up
Org Process Res Dev 2014, 18(12): 1652
https://pubs.acs.org/doi/full/10.1021/op500270z
https://pubs.acs.org/doi/suppl/10.1021/op500270z/suppl_file/op500270z_si_001.pdf
Synthesis of Akt Inhibitor Ipatasertib. Part 2. Total Synthesis and First Kilogram Scale-up

Herein, the first-generation process to manufacture Akt inhibitor Ipatasertib through a late-stage convergent coupling of two challenging chiral components on multikilogram scale is described. The first of the two key components is a trans-substituted cyclopentylpyrimidine compound that contains both a methyl stereocenter, which is ultimately derived from the enzymatic resolution of a simple triester starting material, and an adjacent hydroxyl group, which is installed through an asymmetric reduction of the corresponding cyclopentylpyrimidine ketone substrate. A carbonylative esterification and subsequent Dieckmann cyclization sequence was developed to forge the cyclopentane ring in the target. The second key chiral component, a β2-amino acid, is produced using an asymmetric aminomethylation (Mannich) reaction. The two chiral intermediates are then coupled in a three-stage endgame process to complete the assembly of Ipatasertib, which is isolated as a stable mono-HCl salt.
(S)-2-(4-Chlorophenyl)-1-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-(isopropylamino)propan-1-one, Ipatasertib Mono-HCl
Ipatasertib mono-HCl (3.23 kg, 80% yield) as an off-white solid. Analytical results: 99.7 A% [0.26% S,R,S-diastereomer observed)]; impurity 23 (M399) was not detected (<0.02 A%) [Method 2.2]; ruthenium content by IPC-AES = 5 ppm; analysis for PF6 anion by CAD-HPLC resulted in not detected [Method 2.3]; residual solvent = 0.4% EtOAc; ion chromatography (IC) = 8.5% chloride (1.14 salt equivalent); DSC = 141 °C; FTIR (neat) 3269 (br OH), 2961–2865 (N–H stretch), 1637 (C═O stretch); 1H NMR (600 MHz, DMSO-d6) 9.39 (s, 1H), 8.64 (s, 1H), 8.49 (s, 1H), 7.49 (q, J = 2.9 Hz, 2H), 7.41 (q, J = 2.9 Hz, 2H), 5.58 (s, 1H), 4.91 (t, J = 6.9 Hz, 1H), 4.78 (dd, J = 8.9, 4.5 Hz, 1H), 3.81 (m, J = 3.3 Hz, 1H), 3.68 (m, J = 3.3 Hz, 1H), 3.67 (m, J = 3.1 Hz, 1H), 3.65 (m, J = 3.2 Hz, 1H), 3.63 (m, J = 3.6 Hz, 1H), 3.59 (m, J = 4.3 Hz, 1H), 3.51 (m, J = 3.5 Hz, 1H), 3.46 (m, J = 3.5 Hz, 1H), 3.36 (m, J = 3.2 Hz, 1H), 3.30 (m, J = 5.7 Hz, 1H), 3.21 (m, J = 3.4 Hz, 1H), 2.98 (m, J = 5.8 Hz, 1H), 1.97 (m, J = 4.8 Hz, 2H), 1.26 (d, J = 6.6 Hz, 3H), 1.25 (d, J = 7.0 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) 170.2, 168.2, 159.4, 155.2, 135.3, 132.5, 129.7 (2C), 129.1 (2C), 120.8, 71.7, 50.4, 47.0, 44.8, 44.5, 44.1, 41.4, 40.8, 34.5, 19.8, 18.4, 18.1; HRMS calcd for C24H32ClN5O2 457.2245; found [M+H]+ 458.2306.
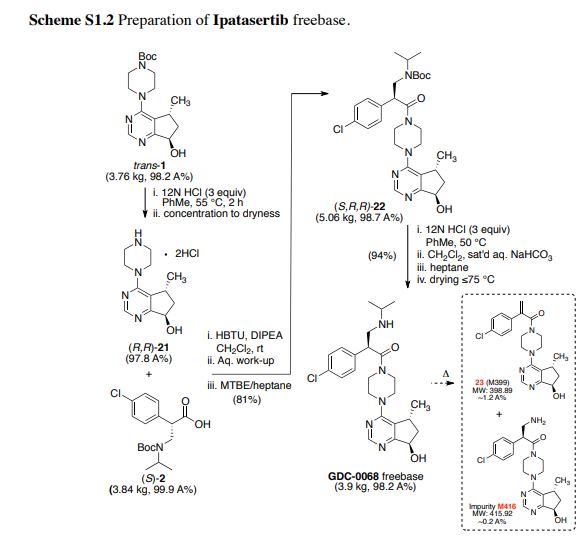
Ipatasertib freebase (3.9 kg, 98.2 A% containing ~1.2% impurity 23 (M399) and impurity M416 at 0.2 A% [Method 2.2]) as tan solid. By CAD-HPLC (see Figure S1-2), the PF6 anion was present in ~0.86 A% [Method 2.3]; Ion chromatography (IC) = 4.0% chloride (0.56 salt equivalent); 1 H NMR (600 MHz, DMSO-d6) 8.44 (s, 1H), 7.45 (d, J = 8.5 Hz, 2H), 7.40 (d, J = 8.5 Hz, 2H), 5.48 (br s, 1H), 4.86 (t, J = 6.9 Hz, 1H), 4.58 (dd, J = 7.3, 4.6 Hz, 1H), 3.74 (m, 1H), 3.40 (m, 1H), 3.63 (m, 2H), 3.61 (m, 1H), 3.42 (m, 1H), 3.57 (m, 1H), 3.18 (m, 1H), 3.50 (m, J = 2.9 Hz, 1H), 3.09 (m, J = 3.1 Hz, 1H), 3.42 (m, 1H), 2.87 (m, J = 4.7 Hz, 1H), 2.00 (m, 1H), 1.92 (m, J = 3.1 Hz, 1H), 1.15 (d, J = 6.4 Hz, 6H), 1.03 (d, J = 6.9 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) 172.0, 169.0, 159.6, 156.3, 136.3, 132.1, 129.7 (2C), 128.9 (2C), 120.9, 72.0, 49.4, 48.7, 45.4, 44.9, 44.8, 44.6, 41.4, 40.9, 34.3, 20.1, 19.9, 19.7; HRMS calcd for C24H32ClN5O2 [M+H]+ 458.2317; found 458.2312. See supporting information (S2) for the NMR spectra (DMSO-d6) of Ipatasertib freebase: ( 1 H) S2, Figure S2-5.12 and ( 13C) Figure S2-5.13.
https://pubs.acs.org/doi/suppl/10.1021/op500270z/suppl_file/op500270z_si_002.pdf
Table S2-1 1 H NMR Assignments of Ipatasertib mono-HCl. S2-52 Figure S2-5.10. 13C NMR (DMSO-d6) spectrum of Ipatasertib mono-HCl. S2-53 Table S2-2 13C NMR Assignments of Ipatasertib mono-HCl. S2-54 Table S2-3 Characteristic Ipatasertib mono-HCl Infrared Signals. S2-55 Figure S2-5.11. FTIR Spectrum of Ipatasertib mono-HCl. S2-56 Figure S2-5.12. XRPD Pattern of Ipatasertib mono-HCl. S2-57
PAPER
https://pubs.acs.org/doi/abs/10.1021/op500271w
https://pubs.acs.org/doi/suppl/10.1021/op500271w/suppl_file/op500271w_si_001.pdf
Synthesis of Akt Inhibitor Ipatasertib. Part 1. Route Scouting and Early Process Development of a Challenging Cyclopentylpyrimidine Intermediate

Herein, the route scouting and early process development of a key cyclopentylpyrimidine ketone intermediate toward the synthesis of Akt inhibitor Ipatasertib are described. Initial supplies of the intermediate were prepared through a method that commenced with the natural product (R)-(+)-pulegone and relied on the early construction of a methyl-substituted cyclopentyl ring system. The first process chemistry route, detailed herein, enabled the synthesis of the ketone on a hundred-gram scale, but it was not feasible for the requisite production of multikilogram quantities of this compound and necessitated the exploration of alternative strategies. Several new synthetic approaches were investigated towards the preparation of the cyclopentylpyrimidine ketone, in either racemic or chiral form, which resulted in the discovery of a more practical route that hinged on the initial preparation of a highly substituted dihydroxypyrimidine compound. The cyclopentane ring in the target was then constructed through a key carbonylative esterification and subsequent tandem Dieckmann cyclization–decarboxylation sequence that was demonstrated in a racemic synthesis. This proof-of-concept was later developed into an asymmetric synthesis of the cyclopentylpyrimidine ketone, which will be described in a subsequent paper, along with the synthesis of Ipatasertib.
PAPER
Discovery and preclinical pharmacology of a selective ATP-Competitive akt inhibitor (GDC-0068) for the treatment of human tumors
J Med Chem 2012, 55(18): 8110
PAPER
Asymmetric synthesis of akt kinase inhibitor ipatasertib
Org Lett 2017, 19(18): 4806
PATENT
WO 2008006040
PATENT
WO 2012135753
PATENT
WO 2012135759
PATENT
WO 2012135781
PATENT
WO 2013173784
PATENT
WO 2015073739
PATENT
WO 2012135779
PATENT
WO 2013173768
References
- Jump up^ https://www.clinicaltrials.gov/ct2/show/NCT02301988
- Jump up^ Lin K, Friedman L, Gloor S, Gross S, Liederer BM, Mitchell I, et al. Preclinical characterization of GDC-0068, a novel selective ATP competitive inhibitor of Akt. 22nd-EORTC-NCI-AACR-2010 2010; abstr. 79
 |
|
| Clinical data | |
|---|---|
| Routes of administration |
PO |
| ATC code |
|
| Identifiers | |
| ChemSpider | |
| KEGG | |
| Chemical and physical data | |
| Formula | C24H32ClN5O2 |
| Molar mass | 458.00 g·mol−1 |
| 3D model (JSmol) | |
////////////// ipatasertib, orphan drug designation, GDC-0068 , RG7440, PHASE 3
CC(C)
It has been found that ipatasertib exhibits a very high solubility (>1 g/g water; >2 g/g water/ethanol 1:1) and a very high hygroscopicity (˜6% at 50% RH, >35% at 95% RH). Whereas poor solubility is often a limiting factor in the development of galenical formulations of other API’s (active pharmaceutical ingredient), a high solubility can equally be problematic for the process performance. Due to this very high intrinsic hygroscopicity of the API, ipatasertib drug substance tends to auto-dissolve to a honey-like viscous liquid at increased humidity. Such high solubility and hygroscopicity may pose serious problems for processing as well as for stability and shelf-life of the final product. Therefore, conventional pharmaceutical compositions comprising ipatasertib and processes for the manufacture of pharmaceutical compositions comprising wetting (e.g. wet granulation) are difficult due to the high solubility and high hygroscopicity of the API.