














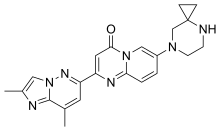
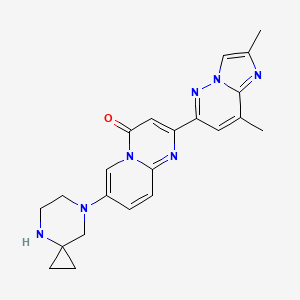
RISDIPLAM
RG-7916, RO-7034067, リスジプラム
| Formula |
C22H23N7O
|
|---|---|
| Cas |
1825352-65-5
|
| Mol weight |
401.4643
|
| US9969754 |
7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[1,2-b]pyridazin-6-yl)pyrido[1,2-a]pyrimidin-4-one
The compound was originally claimed in WO2015173181 , for treating spinal muscular atrophy (SMA). Roche , under license from PTC Therapeutics , and Chugai , are developing risdiplam (RO-7034067; RG-7916), a small-molecule survival motor neuron (SMN)2 gene splicing modulator and a lead from an SMN2 gene modulator program initiated by PTC Therapeutics in collaboration with the SMA Foundation , for the oral treatment of spinal muscular atrophy
The product was granted orphan drug designation in the U.S., E.U. and in Japan for the treatment of spinal muscular atrophy. In 2018, it also received PRIME designation in the E.U. for the same indication.
Risdiplam (RG7916, RO7034067) is a highly potent, selective and orally active small molecule experimental drug being developed by F. Hoffmann-La Roche, PTC Therapeutics and SMA Foundation to treat spinal muscular atrophy (SMA). It is a pyridazine derivative that works by increasing the amount of functional survival of motor neuron protein produced by the SMN2 gene through modifying its splicing pattern.[1][2]
As of September 2018, risdiplam is undergoing late-stage clinical trials across the spectrum of spinal muscular atrophy[3][4][5] where it has shown promising preliminary results.[6][7]
PATENT
WO2015173181
Example 20
7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6- yl)pyrido[l,2-a]pyrimidin-4-one
In a sealed tube, 2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-7-fluoro-pyrido[l,2-a]pyrimidin-4-one (Intermediate 2; 50 mg, 0.162 mmol), DIPEA (0.22 mL, 1.29 mmol, 4 eq.) and 4,7-diazaspiro[2.5]octane dihydrochloride (32 mg, 0.320 mmol, 3.0 eq.) were stirred in
DMSO (2 mL) at 130°C for 48 hours. The solvent was removed under high vacuum. The residue was taken up in CH2CI2 and washed with an aqueous saturated solution of NaHC03. The organic layer was separated and dried over Na2S04 and concentrated in vacuo. The crude was purified by column chromatography (Si02, CH2Cl2/MeOH=98/2 to 95/5) to afford the title product (12 mg, 18%) as a light yellow solid. MS m/z 402.3 [M+H+].
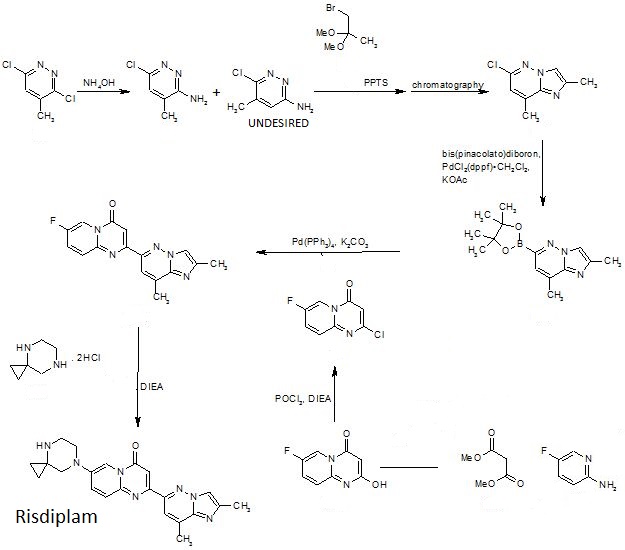
PATENT
WO-2019057740
Process for the preparation of risdiplam and its derivatives.
Scheme 1:
Scheme 3:
Scheme 4:
xample 1: tert-Butyl 7-(6-chloro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate
5-Bromo-2-chloropyridine (85.0 g, 442 mmol), tert-butyl 4,7-diazaspiro[2.5]octane-4-carboxylate (102 g, 442 mmol) and Me-THF (722 g) were charged into a reaction vessel. After 10 minutes stirring, most of the solids were dissolved and [Pd(Xantphos)Cl2] (3.34 g) was added followed after 5 minutes by a solution of sodium tert-butanolate (56.3 g, 574 mmol) in Me-THF (173 g). The reaction mixture was stirred at 70 °C for 1.25 hours, cooled to room temperature and water (595 g) and 1-propylacetate (378 g) were added. After vigorous stirring, the phases were separated, the organic phase was washed with a second portion of water (425 g) and with a mixture of water (425 g) and brine (25 mL). The organic phase was treated with active charcoal (6.8 g), filtered and concentrated under reduced pressure to afford a brown oil, which was dissolved in tert-amyl-methyl-ether (347 g) at reflux. The solution was cooled slowly to room temperature. After stirring 18 hours at room temperature, n-heptane (205 g) was added and the suspension was further cooled to -10 °C. The precipitate was filtered off and dried under high vacuum to afford tert-butyl 7-(6-chloro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (110.9 g, 77.5%) as a beige solid.
Ή-ΝΜΡν (CDC13, 600 MHz): 7.95 (d, 1H); 7.18 – 7.14 (m, 1H); 7.13 – 7.09 (m, 1H); 3.79 – 3.63 (m, 2H); 3.24 – 3.12 (m, 2H); 2.96 (s, 2H); 1.47 (s, 9H); 1.11 – 1.04 (m, 2H); 0.90 -0.79 (m, 2H); LCMS: 324.15, 326.15 (M+H+)
Example 2: tert-butyl 7-(6-amino-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate
An autoclave equipped with an ascending pipe was filled with ammonia (78.7 g, 15 eq; 10 eq are sufficient) at -70 °C. Another autoclave was charged with tert-butyl 7-(6-chloro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (100 g, 309 mmol), sodium tert-butanolate (32.6 g, 340 mmol) and dioxane (800 mL). After 10 minutes stirring at room temperature under Ar, a solution of Pd2(dba)3 (1.41 g, 1.54 mmol) and tBuBrettPhos (1.50 g, 3.09 mmol) in dioxane (180 mL) was added. Thereafter, the connected ammonia vessel was warmed with a warm water bath and the connecting valve was opened. The autoclave was warmed to 30 °C and the reaction mixture stirred 5 hours at this temperature. The ammonia vessel was closed and disconnected. The excess ammonia was washed out of the autoclave with Argon. The reaction solution was poured into a separating funnel, the autoclave washed with ethyl acetate (300 mL) and water (100 mL) and these two solvent portions were added to the separating funnel. The biphasic mixture was further diluted with ethyl acetate (900 mL) and water (1000 mL). After vigorous stirring, the phases were separated. The organic phase was washed with a mixture of water (500 mL) and brine (10 mL). The combined aqueous phases were extracted twice with ethyl acetate (500 mL). The combined organic phases were treated with active charcoal (3.70 g, 309 mmol), filtered and the filtrate was concentrated under reduced pressure to afford a thick brown oil. This oil was dissolved in 1 -propyl acetate (160 mL) at 45-50°C and n-heptane (940 mL) was added drop wise within 1.5 hours. The suspension was cooled slowly to -5°C, stirred 4 hours at -5 °C and filtered. The precipitate was washed with cold n-heptane and dried under high vacuum at 50°C to afford tert-butyl 7-(6-amino-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (81.4 g, 86.5%) as a beige solid.
Ή-ΝΜΡν (CDCb, 600 MHz): 7.71 (d, 1H); 7.12 (dd, 1H); 6.47 (d, 1H); 4.18 (br s, 2H); 3.74 – 3.58 (m, 2H); 3.09 – 2.94 (m, 2H); 2.81 (s, 2H); 1.52 – 1.39 (m, 9H); 1.17 – 0.98 (m, 2H); 0.92 – 0.75 (m, 2H); LCMS: 305.20 (M+H+)
Example 3: tert-butyl 7-(6-amino-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate
An autoclave was charged with tert-butyl 7-(6-chloro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (339 mg, 1 mmol), sodium tert-butanolate (109 mg, 1.1 mmol) and dioxane (5 mL). After 5 minutes stirring at room temperature under Argon [Pd(allyl)(tBuBrettPhos)]OTf (4 mg, 5 μιηοΐ) was added. Thereafter, the autoclave was closed and connected to an ammonia tank, the valve was open and ammonia (230 mg, 13.5 mmol) was introduced into the autoclave. The valve was closed and the autoclave disconnected. The autoclave was warmed to 30 °C and the reaction mixture stirred 4 hours at this temperature. Then the autoclave was opened and the excess ammonia was washed out of the autoclave with Argon. The reaction solution was poured into a flask and taken to dryness under reduced pressure. The residue was purified by chromatography over silica gel (eluent: dichloromethane/ethyl acetate to dichloromethane/methanol). After evaporation of the solvents tert-butyl 7-(6-amino-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (283 mg, 93%) was isolated as a brown oil containing 4% dichloromethane and 3% ethyl acetate.
Example 4: tert-butyl 7-(6-nitro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate
tert-Butyl 4,7-diazaspiro[2.5]octane-4-carboxylate oxalate salt (2.46 kg, 8.13 mol), 5-bromo-2-nitro-pyridine (1.50 kg, 7.39 mol) and dimethyl sulfoxide (7.80 L) were char; into a reaction vessel pre-heated to 35 °C. With stirring, and keeping the temperature below 40°C, lithium chloride (1.25 kg, 25.6 mol) was added portion- wise followed by tetramethylguanidine (2.98 kg, 25.9 mol). Dimethyl sulfoxide (450 mL) was used to rinse the feed line. The reaction mixture was stirred at 79 °C for 8 hours, cooled to 70°C and water (2.48 L) was added within 2 hours. After stirring at 70 °C for an additional 1 hour, the precipitate was filtered off and washed with water (4.5 L) three times. The precipitate was dissolved in ethyl acetate (15 L) and water (7.5 L) at reflux temperature. The phases were separated at 60°C and n-heptane (7.5 L) was added to the organic layer at 60°C within 30 minutes. The solution was cooled to 0°C in 2 hours and further stirred at 0°C for 1 hour. The precipitate was filtered off, washed with a mixture of ethyl acetate (750 mL)/n-heptane (375 mL) twice and dried under reduced pressure to afford 1.89 kg (76.4%) of tert-butyl 7-(6-nitro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate as a yellow to light brown solid.
!H-NMR (CDCls, 600 MHz): 8.16 (d, 1H); 8.07 (d, 1H); 7.15 (dd, 1H); 3.80 – 3.72 (m, 2H); 3.49 – 3.41 (m, 2H); 3.23 (s, 2H); 1.48 (s, 9H); 1.16 – 1.08 (m, 2H); 0.92 – 0.85 (m, 2H); LCMS: 335.17 (M+H+)
Example 5: tert-butyl 7-(2-hydroxy-4-oxo-pyrido[l,2-a]pyrimidin-7-yl)-4,7-diazaspiro[2.5]octane-4-carboxylate
tert-Butyl 7-(6-amino-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (80.0 g, 263 mmol) was dissolved in anisole (800 mL) and di-tert-butyl malonate (71.1 g, 315 mmol) was added. The solution was stirred 3.5 hours at 145 °C then cooled to room temperature. The precipitate was filtered off, washed with toluene (in portions, 320 mL in total) and dried under high vacuum at 50°C to afford tert-butyl 7-(2-hydroxy-4-oxo-pyrido[l,2-a]pyrimidin-7-yl)-4,7-diazaspiro[2.5]octane-4-carboxylate (65.6 g, 67%) as a light pink powder.
Ή-ΝΜΡν (CDCI3, 600 MHz): 8.46 (d, 1H); 7.74 (dd, 1H); 7.52 (d, 1H); 5.37 (s, 2H); 3.83 – 3.69 (m, 2H); 3.23 (t, 2H); 3.01 (s, 2H); 1.48 (s, 9H); 1.17 – 1.03 (m, 2H); 0.95 – 0.75 (m, 2H); LCMS: 373.19 (M+H+)
Example 6: tert-butyl 7-(2-hydroxy-4-oxo-pyrido[l,2-a]pyrimidin-7-yl)-4,7-diazaspiro[2.5]octane-4-carboxylate
tert-Butyl 7-(6-nitro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (950 g, 2.84 mol), Pt 1%, V 2% on active charcoal (95.1 g, 2 mmol) and ethyl acetate (9.5 L) were charged into an autoclave that was pressurized with hydrogen gas to 3 bar. The reaction mixture was stirred at room temperature for 6 hours. The excess hydrogen was vented. The reaction mixture was filtered, the catalyst was washed with ethyl acetate (0.95 L) three times. The filtrate was concentrated under reduced pressure and the solvent exchanged to anisole (add two portions of 2.85 L and 5.18 L) by distillation. Di tert-butyl malonate (921.7 g, 4.26 mol) was added and the charging line was rinsed with anisole (618 mL) and the reaction mixture was stirred at 125-135 °C for 8 hours. It may be necessary to distill off the by-product tert-butanol to reach this temperature. The progress of the reaction was followed eg.by HPLC. If the reaction stalls, the temperature is increased to 135-145°C and checked for progress after 1 hour. When the reaction was complete, the batch was cooled to room temperature and stirred at room temperature for 4 hours. The precipitate was filtered off, washed with toluene (3.55 L) and dried under vacuum at 60°C to afford tert-butyl 7-(2-hydroxy-4-oxo-pyrido[l,2-a]pyrimidin-7-yl)-4,7-diazaspiro[2.5]octane-4-carboxylate (861.0 g, 81.4%) as a yellow to light brown solid.
Example 7: tert-butyl 7-[4-oxo-2-(p-tolylsulfonyloxy)pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate
A reactor was charged with tert-butyl 7-(2-hydroxy-4-oxo-pyrido[l,2-a]pyrimidin-7-yl)-4,7-diazaspiro[2.5]octane-4-carboxylate (920 g, 2.47 mol) and then triethylamine (325 g, 3.21 mol), followed by tosyl chloride (527.1 g, 2.77 mol) and dichloromethane (4.6 L). The reaction mixture was stirred at 20-25 °C for at least three hours. Upon complete reaction, the organic solution was washed with a prepared solution of HC1 (32%, 247.8 mL) and water (4.6 L), followed by a prepared solution of sodium hydroxide (432.3 mL of a 30% stock solution) and water (3.9 L) in that order. The organic phase was finally washed with water (4.8 L) and then dichloromethane was nearly completely distilled off under reduced pressure at 50-55°C. Ethyl acetate (920 mL) was added and distilled twice at this temperature under reduced pressure, and then ethyl acetate (4.8 L) was added and the suspension cooled to 20-25 °C over two hours. n-Heptane (944.4 mL) was added and the mixture was cooled to 0-5 °C and then stirred for an additional 3 hours. The precipitate was filtered off, washed with a prepared solution of ethyl acetate (772.8 mL) and n-heptane (147.2 mL), and then twice with n-heptane (2.6 L). The solid was dried under vacuum at 45-50°C to afford 1122.6 g (86.3%) tert-butyl 7-[4-oxo-2-(p-tolylsulfonyloxy)pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate as yellow crystals.
!H-NMR (CDCls, 600 MHz): 8.32 (d, 1H); 8.00 – 7.89 (m, 2H); 7.66 (dd, 1H); 7.50 (d, 1H); 7.36 (d, 2H); 6.04 (s, 1H); 3.80 – 3.68 (m, 2H); 3.23 (t, 2H); 3.01 (s, 2H); 1.48 (s, 9H); 1.15 – 1.04 (m, 2H); 0.92 – 0.82 (m, 2H); LCMS: 527.20 (M+H+)
Example 8: 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine
6-Chloro-2,8-dimethylimidazo[l,2-b]pyridazine (40.0 g, 220 mmol), bis pinacol diborane (69.9 g, 275 mmol) and potassium acetate (43.2 g, 440 mmol) were suspended in acetonitrile (440 mL). The suspension was heated to reflux and stirred 30 minutes at reflux, then a suspension of PdCl2(dppf) (4.03 g, 5.51 mmol) and dppf (610 mg, 1.1 mmol) in acetonitrile (40 mL) was added. The vessel was rinsed with acetonitrile (20 mL), which were also poured into the reaction mixture. The orange suspension was further stirred at reflux, whereby acetonitrile (50 mL) were distilled off. After 4 hours, the reaction mixture was filtered off, the filter was washed with several portions of acetonitrile (in total 150 mL). The filtrate was diluted to obtain a volume of 700 mL. The 314 mmolar solution of 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine in acetonitrile was used as such in the next step.
Example 9: 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine
6-chloro-2,8-dimethylimidazo[l,2-b]pyridazine (29.0 g, 22.8 mmol), bis pinacol diborane (44.6, 25.1 mmol) and potassium acetate (31.3 g, 45.6 mmol) were suspended in 1-propyl acetate (365 mL). The suspension was heated to 80°C and a solution of
tricyclohexylphosphine (448 mg, 0.23 mmol) and Pd(OAc)2 (179 mg, 0.11 mmol) in 1-propyl acetate (37 mL) was added within 20 minutes. After 2.5 hours further stirring at 80°C, the suspension was cooled to 40°C and filtered at this temperature. The precipitate was washed with 1-propyl acetate (200 mL). The filtrate corresponds to 516.4 g of a 8.5% solution of 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine in 1 -propyl acetate.
Example 10: Isolation of 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[ 1 ,2-b]pyridazine
In another experiment, the above solution obtained was cooled to 0-5 °C within 3 hours. The precipitate was filtered off, washed with cold 1 -propyl acetate and dried under high vacuum at 60°C to afford 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine (24. Og, 55%) as a colourless solid.
lH NMR (CDCls, 600 MHz, ) δ ppm 7.86 (d, J=0.7 Hz, 1 H), 7.20 (d, J=1.0 Hz, 1 H), 2.63 (d, J=1.0 Hz, 3 H), 2.51 (d, J=0.7 Hz, 3 H), 1.33 – 1.49 (m, 12 H)
Example 11: (step 6) tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate
tert-Butyl 7-[4-oxo-2-(p-tolylsulfonyloxy)pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5] octane-4-carboxylate (25 g, 47.5 mmol), 2,8-dimethyl-6-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine (314 mM in acetonitrile, 191 mL, 59.8 mmol), PdCi2(dppf) (868 mg, 1.19 mmol) and aqueous potassium carbonate 4.07 M (17.1 mL, 69.8 mmol) were charged into a reaction vessel. The reaction mixture was stirred at reflux for 3 hours, cooled overnight to room temperature and filtered. The precipitate was washed with several portions of acetonitrile (146 mL in total), then suspended in methyl-THF (750 mL) and methanol (75 mL). Aqueous sodium hydrogen carbonate 5% (250 mL) was added, the mixture was vigorously stirred at 35°C. The phases were separated, the organic phase was washed again with aqueous sodium hydrogen carbonate 5% (250 mL). The organic phase was treated with active charcoal for 1 hour at room temperature, filtered and the filtrate was concentrated under reduced pressure at 60 °C to a volume of 225 mL, heated to reflux then cooled to room temperature, stirred at room temperature for 16 hours, then cooled to 0°C and stirred at 0°C for 3 hours. The precipitate was filtered off, washed with n-heptane (60 mL) and dried under high vacuum at 55°C to afford tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (20.13 g, 84.5%) as a yellow solid.
This solid could be recrystallized in the following manner: 15 g of the above solid was dissolved at reflux in toluene (135 mL) and ethanol (15 mL). The solution was slowly cooled to room temperature, stirred 16 hours at room temperature, then cooled to 0°C and stirred at 0°C for 4 hours. The precipitate was filtered off, washed with cold toluene and dried under high vacuum at 55°C to afford tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (11.92 g, 79.5%) as a yellow-green solid.
!H-NMR (CDCls, 600 MHz): 8.44 (d, 1H); 7.93 (d, 1H); 7.96 – 7.89 (m, 1H); 7.80 (d, 1H); 7.76 – 7.72 (m, 1H); 7.70 – 7.63 (m, 1H); 7.38 (s, 1H); 3.85 – 3.69 (m, 2H); 3.28 (t, 2H); 3.07 (s, 2H); 2.74 (d, 3H); 2.55 (s, 3H); 1.49 (s, 9H); 1.16 – 1.09 (m, 2H); 0.93 – 0.86 (m, 2H); LCMS: 502.26 (M+H+)
Example 12: tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate
6-chloro-2,8-dimethylimidazo[l,2-b]pyridazine (4.14 g, 22.8 mmol), bis pinacol diborane (6.37g, 25.1 mmol) and potassium acetate (4.47 g, 45.6 mmol) were suspended in 1-propyl acetate (59 mL). The suspension was heated to 80°C and a solution of
tricyclohexylphosphine (63.9 mg, 0.23 mmol) and Pd(OAc)2 (25.6 mg, 0.11 mmol) in 1-propyl acetate (6 mL) was added within 20 minutes. After 2.5 hours further stirring at 80°C, the suspension was cooled to 40°C and filtered at this temperature. The precipitate was washed with 1-propyl acetate (32 mL). The filtrate corresponds to 74.6 g of a 8.5% solution of 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine in 1-propyl acetate.
A reaction vessel was charged with tert-butyl 7-[4-oxo-2-(p-tolylsulfonyloxy)pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (10.0 g, 19.0 mmol), tricyclohexylphosphine (58.6 mg, 0.21 mmol) and Pd(OAc)2 (21.3 mg, 0.10 mmol) and 1-propyl acetate (42 mL) and a solution of potassium carbonate (5.25 g, 38.0 mmol) in water (19.0 mL) was added. The suspension was heated to 70°C and the solution of 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine in 1-propyl acetate was added within 30 minutes. The mixture was stirred for 2 hours at 70-75°C. The suspension was cooled to 40°C, water (10 mL) was added. The suspension was aged for 30 minutes. The crude product was filtered off and rinsed with 1-propyl acetate (41 mL). The crude product was taken up in toluene (100 mL), 5% aqueous NaHC03-solution (30 mL) and 1-propanol (20.0 mL). The mixture was heated to 60-65 °C, the phases were separated and the organic phase was washed with 2 more portions of water (30.0 mL). The organic phase was filtered on active charcoal, the filter washed with toluene (60.0 mL). The filtrate was concentrated under reduced pressure to a volume of ca. 120 mL, heated to reflux and 1-propanol (0.8 mL) was added to obtain a solution. The solution was cooled to 0-5°C within 4-6 hours, stirred at 0-5°C for 1 hour. The precipitate was filtered off, washed with toluene (30 mL) and dried under reduced pressure at 70-80°C to afford tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (7.7 g, 80.8%) as a yellowish solid.
Example 13: 7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)pyrido[l,2-a]pyrimidin-4-one di-hydrochloride salt
To prepare a solution of HC1 in in 1-propyl acetate/ 1-propanol, acetyl chloride (15.8 g, 199 mmol) was slowly added to a mixture of 1-propyl acetate (60 mL) and 1-propanol (30 mL) at 0°C, and stirring was pursued for an additional 2 hours at room temperature.
tert-Butyl 7-[2-(2,8-dimethylimidazo[ 1 ,2-b]pyridazin-6-yl)-4-oxo-pyrido[ 1 ,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (20 g, 39.9 mmol) was suspended in 1-propyl acetate (60 mL) and 1-propanol (30 mL) at room temperature and the HC1 solution in 1-propyl acetate and 1-propanol was added. The reaction mixture was heated within 3 hours to 70°C and stirred 16 hours at this temperature, then cooled to 20°C. The precipitate was filtered off, washed with 1-propyl acetate (50 mL) in several portions and dried under vacuum at 55 °C to afford 7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)pyrido[l,2-a]pyrimidin-4-one hydrochloride salt (18.8 g, 99%) as yellow crystals.
^-NMR (CDCls, 600 MHz): 8.34 (s, 1H); 8.22(s, 1H); 8.05 (s, 1H); 8.01 (dd, 1H); 7.80 (d, 1H); 7.16 (s, 1H); 3.71 – 3.67 (m, 2H); 3.64 – 3.59 (m, 2H); 3.52 (s, 2H); 2.69 (s, 3H); 2.54 (s, 3H); 1.23- 1.20 (m, 2H); 1.14 – 1.08 (m, 2H); LCMS: 402.20 (M+H+)
Example 14: 7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)pyrido[ 1 ,2-a]pyrimidin-4-one
To a suspension of tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (25 g, 50 mmol) in 1-propyl acetate (375 mL) was added a solution of HC1 in 1-propanol (prepared by adding slowly at 5°C acetyl chloride (18.0 mL) to 1-propanol (37.6 mL) and stirring 1 hour at room temperature). The stirred suspension was heated to 75°C within 10 hours and stirred a further 5 hours at 75 °C. Water (160.0 mL) was added and the phases were separated at 75°C. Aqueous sodium hydroxide 32% (27.8 mL) was added to the aqueous phase. The suspension obtained was cooled to room temperature within 5 hours and stirred one hour at room temperature. The precipitate was filtered off, washed with water (100.0 mL) and dried under reduced pressure at 50°C for 18 hours to afford 7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)pyrido[l,2-a]pyrimidin-4-one (19.7 g, 98.3%) as yellow crystals.
!H-NMR (CDCb, 600 MHz): 8. 45 (d, 1H); 7.92 (d, 1H); 7.80 (s, 1H); 7.75 – 7.71 (m, 1H); 7.71 – 7.67 (m, 1H); 7.37 (s, 1H); 3.31 – 3.24 (m, 2H); 3.22 – 3.16 (m, 2H); 3.09 (s, 2H); 2.73 (s, 3H); 2.55 (s, 3H); 0.82- 0.76 (m, 2H); 0.71 – 0.63 (m, 2H); LCMS: 402.20
(M+H+)
Example 15: 7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)pyrido[ 1 ,2-a]pyrimidin-4-one
A suspension of tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (13.5 g, 26.9
in toluene (237.0 g) was stirred at 75°C and a 21.9% solution of HCl in 1-propanol (21.4 g, 134.5 mmol) was added within 2.5 hours. The reaction mixture was stirred further at 75 °C until complete conversion. The reaction mixture was cooled to 20-25°C. Water (70 g) was added. The biphasic mixture was stirred another 10 minutes at 20-25 °C and the phases were separated. The organic phase was extracted with water (17 g) twice and the combined aqueous phases were added into mixture of aqueous sodium hydroxide 28% (15.0 g) and water (45.0 g). The suspension obtained was cooled to 20°C. The precipitate was filtered off , washed with water (25 g) three times and dried under reduced pressure at 60°C to afford 7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)pyrido[l,2-a]pyrimidin-4-one (9.5 g, 95.1%) as yellow crystals.
Example 16: 4-bromo-6-chloro-pyridazin-3-amine
3-amino-6-chloropyridazine (20 g, 154 mmol), sodium bicarbonate (25.9 g, 309 mmol) and methanol (158 g) were charged in a reaction vessel and cooled to 0-10°C. Bromine (34.5 g, 216 mmol) was added drop wise and the reaction mixture was stirred 3 days at room temperature. 10% Aqueous sodium sulfate was added. The suspension was filtered off. The filtrate was washed with ethyl acetate (300 mL) twice. The combined organic layers were dried and evaporated. A suspension of the residue in methanol (50 mL) was heated to reflux, water (120 mL) was added and the suspension was stirred 16 hours at room temperature. The precipitate was filtered off and dried. The residue was suspended in n-heptane (50 mL), stirred 2 hours at room temperature, filtered off and dried to afford 4-bromo-6-chloro-pyridazin-3-amine (14.5 g, 46.2%) as a light brown solid.
!H-NMR (CDCls, 600 MHz): 7.55 (s, 1H); 5.83-4.89 (m, 2H); LCMS: 209.93 (M+H+)
Example 17: 4-bromo-6-chloro-pyridazin-3-amine
3-amino-6-chloropyridazine (50 g, 360 mmol), acetic acid (5.8 g, 96.5 mmol), sodium acetate (28.7 g, 289.5 mmol) and methanol (395 g) were charged in a reaction vessel and heated to 25-35°C. Dibromodimethylhydatoin (66.0 g, 231.6 mmol) was added in several portions and the reaction mixture was stirred 3 hours at 30°C. Completion is checked by IPC and if the conversion is incomplete, dibromodimethylhydantoin is added (5.5g). At reaction completion, 38% aqueous sodium sulfate (77.2 mmol NaHS03) was added slowly. The suspension was concentrated under reduced pressure and water (500 g) was added slowly at 45°C, then 30% aqueous sodium hydroxide (31.5 g, 231.6 mmol NaOH) was added at 20°C to adjust pH to 7-8. The precipitate was filtered off, washed with water and dried under reduced pressure to afford 4-bromo-6-chloro-pyridazin-3-amine (50.2 g, 62.5%) as a grey solid.
Example 18: 6-chloro-4-methyl-pyridazin-3-amine
4-bromo-6-chloro-pyridazin-3-amine (3.0 g, 14.4 mmol) and
tetrakis(triphenylphosphine)palladium (1666 mg, 144 μιηοΐ) were suspended in THF (13.2 g) and a solution of zinc chloride in Me-THF (2.0 M, 9 mL, 18 mmol) was added. The reaction mixture was cooled to -5°C and methyllithium in diethoxymethane (3.1 M, 11.6 mL, 36 mmol) was added. The reaction mixture was stirred at 45°C for 4 hours. Sodium sulfate decahydrate (11.7 g, 36 mmol) was added at room temperature, the mixture was stirred 1.5 hours at 60°C, diluted with water (100 mL) and after 30 minutes the precipitate was filtered off. The precipitate was dissolved in aqueous HC1 2M (100 mL) and ethyl acetate (140 mL). The biphasic system was filtered, the phases were separated and the pH of the water layer adjusted to 7 with aqueous NaOH 32% (18 mL). The precipitate was filtered and dried. The solid obtained was digested twice in methanol (20 mL) at room temperature. The two filtrates were combined, evaporated and dried under high vacuum to afford 6-chloro-4-methyl-pyridazin-3-amine (1.2 g, 58.1%) as a red solid.
Ή-ΝΜΡν (CDCb, 600 MHz): 7.09 (d, 1H); 4.90 (br s, 2H), 2.17 (d, 3H)
Example 19: 6-chloro-4-methyl-pyridazin-3-amine
4-bromo-6-chloro-pyridazin-3-amine (30.02 g, 143 mmol) and THF (180 mL) were charged into a reaction vessel. Methylmagnesium chloride (22% in THF, 50.0 mL, 1.03 eq.) was added at 20°C over 60 minutes, followed by zinc chloride in Me-THF (25%, 37 mL, 0.50 eq.) and palladium tetrakis(triphenyphosphine) (1.66 g, lmol%). The reaction mixture was heated to 50°C and methylmagnesium chloride (22% in THF, 81 mL, 1.7 eq.) was added slowly. The reaction mixture was stirred at 50°C until complete conversion, then at 10°C for 14.5 hours and poured into a mixture of water (90 g), aqueous HCl 33% (52.5 g) and toluene (150 mL) maintained at 20-30°C. The aqueous phase was separated and the organic phase was extracted with a solution of aqueous HCl 33% (2.0 g) and water (45 g). The aqueous layers were combined and washed with toluene (30 mL) twice and the pH was adjusted by addition of 25% aqueous ammonia solution. When a pH of 2.4 was reached, seeding crystals were added, the mixture was stirred further for 15 minutes and thereafter the pH was brought to 4.0. The suspension was stirred at 20°C for 2 hours, the precipitate was filtered off, washed with water (20 mL) three times to afford crude 6-chloro-4-methyl-pyridazin-3-amine (29 g) as a brown solid.
29 g crude product was transferred to a reaction vessel and methanol (20 mL) was added. The mixture was refluxed for 30 minutes and 12 g water was added. The solution was cooled to 0°C and stirred for 2 hours at this temperature. The precipitate was filtered off, washed with water three times and dried under reduced pressure at 40°C to afford purified 6-chloro-4-methyl-pyridazin-3-amine (13.8 g, 66%) as a light brown solid.
Alternative purification:
50 g crude 6-chloro-4-methyl-pyridazin-3-amine were dissolved in methanol (250 mL) and active charcoal (4.0 g) and diatomaceous earth (2.5 g) were added. The suspension was stirred at 45°C for 1 hour, cooled to 30°C and potassium hydrogenophosphate (2.1 g) was added. The suspension was stirred at 30°C for another 90 minutes, filtered and the precipitate washed with methanol (100 mL). The filtrate was concentrated to a residual volume of 175 mL and water (120 mL) was added. The resulting suspension was heated
to reflux affording a solution which was cooled to 20°C resulting in a suspension. The precipitate was filtered off, washed with water (90 mL) and dried under reduced pressure to afford pure 6-chloro-4-methyl-pyridazin-3-amine (38 g, 76%) as a light yellow solid.
Example 20: 6-chloro-2,8-dimethyl-imidazo[l,2-b]pyridazine
6-chloro-4-methyl-pyridazin-3-amine (70.95 kg, 494.2 mol), sodium bromide (35 kg, 345.9 mol), isopropyl acetate (611 kg), isopropanol (28 kg and water (35 kg) were charged into a reaction vessel. The reaction mixture was stirred at 80-85 °C for 8 hours. Isopropyl acetate (310 kg) and water (420 kg) were added. 30% Aqueous NaOH was added at 45-55 °C and the system was stirred for 2 hours. The phases were separated at 25-35 °C. The organic layer was washed with water (370 kg), filtered on diatomite (7 kg) and the filter washed with isopropyl acetate (35 kg). The organic phase was extracted with two portions of 5.4% aqueous sulfuric acid (910 kg followed by 579 kg). The combined aqueous phases were basified with 30% aqueous NaOH (158 kg). The suspension was stirred 2 hours at 15-25 °C. The precipitate was isolated by centrifugation in three portions, each washed with water (31 kg). The wet solid was dissolved in isopropyl acetate (980 kg) at 25-35 °C, the solution washed with water (210 kg), three times. The organic phase was treated with active charcoal for 12 hours at 45-50 °C, concentrated to ca. 300 kg and heated to 70-80 °C to obtain a clear solution. This solution was cooled to 50-60 °C, stirred at this temperature for 1 hour, n-heptane (378 kg) was added and stirring was pursued for 1 hour. The mixture was cooled to -10- -5°C and stirred for another 3 hours. The precipitate was isolated by centrifuging, washed with n-heptane (33 kg) and dried under reduced pressure at 30-50 °C for 15 hours to afford 67.4 kg (76%) 6-chloro-2,8-dimethyl-imidazo[l,2-b]pyridazine as an off-white solid.
XH-NMR (CDCls, 600 MHz): 7.67 (s, 1H); 6.86 (s, 1H); 2.65 (s, 3H), 2.50 (s, 3H)
Paper
https://pubs.acs.org/doi/pdf/10.1021/acs.jmedchem.8b00741

SMA is an inherited disease that leads to loss of motor function and ambulation and a reduced life expectancy. We have been working to develop orally administrated, systemically distributed small molecules to increase levels of functional SMN protein. Compound 2 was the first SMN2 splicing modifier tested in clinical trials in healthy volunteers and SMA patients. It was safe and well tolerated and increased SMN protein levels up to 2-fold in patients. Nevertheless, its development was stopped as a precautionary measure because retinal toxicity was observed in cynomolgus monkeys after chronic daily oral dosing (39 weeks) at exposures in excess of those investigated in patients. Herein, we describe the discovery of 1 (risdiplam, RG7916, RO7034067) that focused on thorough pharmacology, DMPK and safety characterization and optimization. This compound is undergoing pivotal clinical trials and is a promising medicine for the treatment of patients in all ages and stages with SMA.
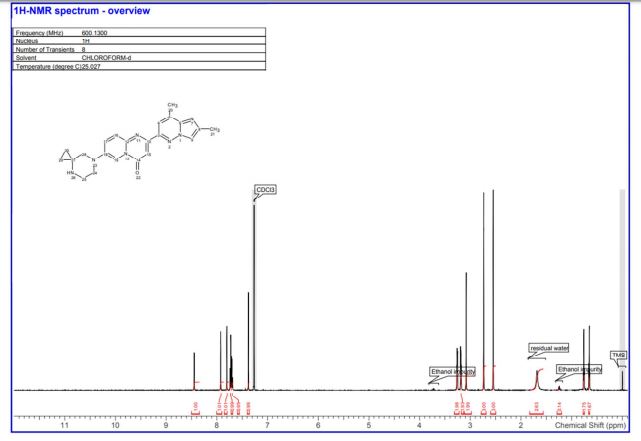
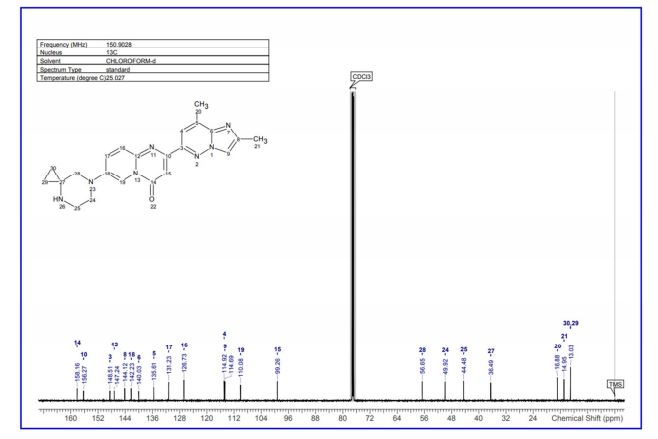
7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[1,2-b]pyridazin-6-yl)pyrido[1,2-a]pyrimidin-4-one 1 (12 mg, 18%) as a pale yellow solid. 1H NMR (600 MHz,CDCl3) δ ppm 8.45 (d, J = 2.4 Hz, 1H), 7.92 (d, J = 1.0 Hz, 1H), 7.73 (d, J = 9.6 Hz, 1H) 7.80 (s, 1H), 7.70 (dd, J = 9.7, 2.5 Hz, 1H), 7.38 (s, 1H), 3.31–3.22 (m, 2H), 3.20–3.16 (m, 2H), 3.08 (s, 2H), 2.74 (d, J = 0.9 Hz, 3H) 2.55 (s, 3H), 1.68 (br s, 1H), 0.77–0.75 (m, 2H), 0.67–0.64 (m, 2 H);
13C NMR (151 MHz,CDCl3) δ ppm 158.2, 156.3, 148.5, 147.2, 144.1, 142.2, 140.0, 135.6, 131.2, 126.7, 114.9, 114.7, 110.1, 99.3, 56.7, 49.9, 44.5, 36.5, 16.9, 15.0, 13.0. LC–HRMS: m/z = 402.2051 [(M + H)+ calcd for C22H24N7O, 402.2042; Diff 0.9 mDa].
References
- ^ Maria Joao Almeida (2016-09-08). “RG7916”. BioNews Services. Retrieved 2017-10-08.
- ^ Zhao, Xin; Feng, Zhihua; Ling, Karen K. Y; Mollin, Anna; Sheedy, Josephine; Yeh, Shirley; Petruska, Janet; Narasimhan, Jana; Dakka, Amal; Welch, Ellen M; Karp, Gary; Chen, Karen S; Metzger, Friedrich; Ratni, Hasane; Lotti, Francesco; Tisdale, Sarah; Naryshkin, Nikolai A; Pellizzoni, Livio; Paushkin, Sergey; Ko, Chien-Ping; Weetall, Marla (2016). “Pharmacokinetics, pharmacodynamics, and efficacy of a small-molecule SMN2 splicing modifier in mouse models of spinal muscular atrophy”. Human Molecular Genetics. 25 (10): 1885. doi:10.1093/hmg/ddw062. PMC 5062580. PMID 26931466.
- ^ “Genentech/Roche Releases Clinical Trial Update for RG7916”. CureSMA. 2017-09-15. Retrieved 2017-10-08.
- ^ “A Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of RO7034067 in Infants With Type1 Spinal Muscular Atrophy (Firefish)”.
- ^ “A Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of RO7034067 in Type 2 and 3 Spinal Muscular Atrophy Participants (Sunfish)”.
- ^ “Updated Preliminary Data from SMA FIREFISH Program in Type 1 Babies Presented at the CureSMA Conference”. www.prnewswire.com. Retrieved 2018-09-11.
 |
|
| Clinical data | |
|---|---|
| Synonyms | RG7916; RO7034067 |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C22H23N7O |
| Molar mass | 401.474 g/mol g·mol−1 |
| 3D model (JSmol) | |
///////////RISDIPLAM, RG-7916, RO-7034067, リスジプラム , PHASE 3, PRIME designation, ORPHAN DRUG
76RS4S2ET1 (UNII code)
CC1=CC(=NN2C1=NC(=C2)C)C3=CC(=O)N4C=C(C=CC4=N3)N5CCNC6(C5)CC6