















ROLAPITANT HYDROCHLORIDE
- Rolapitant HCl
- Rolapitant hydrochloride
- Sch 619734
- SCH619734
- UNII-57O5S1QSAQ
(5S ,8S)-8-[[(1R)-1-[3 ,5-
Bis(trifluoromethyl)phenyl] ethoxy] methyl]-8-phenyl-1,7-
diazaspiro[4.5]decan-2-one hydrochloride monohydrate.
CAS 914462-92-3
Empirical Formula: C25H26F6N2O2 · HCl · H2O, Molecular Weight: 555
USAN Name: Rolapitant hydrochloride, INN Name: rolapitantum or rolapitant
CAS Number: 552292-08-7 (rolapitant free base); 914462-92-3 (rolapitant HCl monohydrdate).
Rolapitant
- Molecular FormulaC25H26F6N2O2
- Average mass500.477 Da
Varubi® is available as tablet for oral use, containing 90 mg of free Rolapitant. The recommended dose is 180 mg approximately 1 to 2 hours prior to the start of chemotherapy.
It is in late-stage trials of its drug rolapitant, which showed promising mid-stage results in reducing nausea and vomiting in patients undergoing chemotherapy
Rolapitant hydrochloride is a tachykinin neurokinin 1 (NK1) antagonist in phase III clinical trials at Tesaro for the prevention of chemotherapy-induced nausea and vomiting (CINV). Phase II clinical trials are also under way at OPKO for this indication. At Merck & Co., phase II clinical studies were also under way for the treatment of chronic idiopathic cough and for the prevention of chemotherapy-induced nausea; however, no recent developments have been reported for these indications.
NK1 is a G-protein coupled receptor found in the central and peripheral nervous systems. Substance P is the endogenous ligand for this receptor, whose activation leads to the production of inositol triphosphate. NK1 is believed to be involved in the emetic response.
The drug candidate was originally developed by Schering-Plough (now Merck & Co.), and in 2009 it was licensed to OPKO for the prevention of nausea and vomiting related to cancer chemotherapy and surgery. In 2010, rolapitant was licensed by OPKO to Tesaro on a worldwide basis for the prevention of chemotherapy-induced nausea and vomiting.
Rolapitant is a selective, bioavailable, CNS penetrant neurokinin NK1 receptor antagonist that shows behavioral effects in animals models of emesis. In vitro studies indicate that rolapitant has a high affinity for the human NK1 receptor of 0.66 nM and high selectivity over the human NK2 and NK3 subtypes of >1000-fold. Rolapitant is a functionally competitive antagonist, as measured by calcium efflux, with a calculated Kb of 0.17 nM. (source: Pharmacol Biochem Behav.2012 Mar 31.
Rolapitant is a potent, selective NK1 receptor antagonist that is rapidly absorbed, has a remarkably long half-life (up to180 hours), and appears to have a low potential for drug-drug interactions. A randomized, multicenter, double-blind, dose-ranging study of rolapitant was conducted with placebo and active control groups. Six hundred nineteen adult women undergoing open abdominal surgery were randomly assigned in equal ratios to 1 of 6 study arms: oral rolapitant in 5-mg, 20-mg, 70-mg, or 200-mg doses; IV ondansetron 4 mg; or placebo, stratified by history of PONV or motion sickness. The primary study endpoint was absence of emetic episodes, regardless of use of rescue medication, at 24 hours after extubation.RESULTS: Groups assigned to rolapitant 20-mg, 70-mg, and 200-mg had a higher incidence of no emesis in comparison with placebo at 24 hours after surgery. A linear relationship between rolapitant dose and primary outcome was seen. The probability of an emetic episode was significantly lower in the rolapitant 70-mg and 200-mg groups in comparison with placebo (P ≤ 0.001 based on the log-rank test). No significant differences were noted between rolapitant and the active control (ondansetron) at 24 hours after surgery, but there was a higher incidence of no emesis (regardless of rescue medication use) in the rolapitant 200- and 70-mg groups at 72 and 120 hours, respectively. CONCLUSION: Rolapitant is superior to placebo in reducing emetic episodes after surgery and reduces the incidence of vomiting in a dose-dependent manner. No differences in side effect profile were observed between rolapitant and placebo.
Rolapitant (INN,[2] trade name Varubi /vəˈruːbi/ və-ROO-bee in the US and Varuby in Europe) is a drug originally developed by Schering-Plough and licensed for clinical development by Tesaro, which acts as a selective NK1 receptor antagonist (antagonist for the NK1 receptor).[3] It has been approved as a medication for the treatment of chemotherapy-induced nausea and vomiting (CINV) after clinical trials showed it to have similar or improved efficacy and some improvement in safety over existing drugs for this application.[4][5][6][7
Medical uses
Rolapitant is used in combination with other antiemetic (anti-vomiting) agents in adults for the prevention of delayed nausea and vomiting associated with initial and repeat courses of emetogenic cancer chemotherapy, including, but not limited to, highly emetogenic chemotherapy.[1] The approved antiemetic combination consists of rolapitant plus dexamethasone and a 5-HT3 antagonist.[8]
Contraindications
Under the US approval, rolapitant is contraindicated in combination with thioridazine, whose inactivation could be inhibited by rolapitant.[9] Under the European approval, it is contraindicated in combination with St. John’s Wort, which is expected to accelerate inactivation of rolapitant.[8]
Side effects
In studies comparing chemotherapy plus rolapitant, dexamethasone and a 5-HT3 antagonist to chemotherapy plus placebo, dexamethasone and a 5-HT3 antagonist, most side effects had comparable frequencies in both groups, and differed more between chemotherapy regimens than between rolapitant and placebo groups. Common side effects included decreased appetite (9% under rolapitant vs. 7% under placebo), neutropenia (9% vs. 8% or 7% vs. 6%, depending on the kind of chemotherapy), dizziness (6% vs. 4%), indigestion and stomatitis (both 4% vs. 2%).[9]
Overdose
Up to eightfold therapeutic doses have been given in studies without problems.[8]
Interactions
Rolapitant moderately inhibits the liver enzyme CYP2D6. Blood plasma concentrations of the CYP2D6 substrate dextromethorphanhave increased threefold when combined with rolapitant; and increased concentrations of other substrates are expected. The drug also inhibits the transporter proteins ABCG2 (breast cancer resistance protein, BCRP) and P-glycoprotein (P-gp), which has been shown to increase plasma concentrations of the ABCG2 substrate sulfasalazine twofold and the P-gp substrate digoxin by 70%.[8]
Strong inducers of the liver enzyme CYP3A4 decrease the area under the curve of rolapitant and its active metabolite (called M19); for rifampicin, this effect was almost 90% in a study. Inhibitors of CYP3A4 have no relevant effect on rolapitant concentrations.[8]
Pharmacology
Pharmacodynamics
Both rolapitant and its active metabolite M19 block the NK1 receptor with high affinity and selectivity: to block the closely related receptor NK2 or any other of 115 tested receptors and enzymes, more than 1000-fold therapeutic concentrations are necessary.[10]
Pharmacokinetics

The major active metabolite, M19 (C4-pyrrolidine-hydroxylated rolapitant).[8] The stereochemistry of the hydroxyl group is unknown.
Rolapitant is practically completely absorbed from the gut, independently of food intake. It undergoes no measurable first-pass effect in the liver. Highest blood plasma concentrations are reached after about four hours. When in the bloodstream, 99.8% of the substance are bound to plasma proteins.[8]
It is metabolized by the liver enzyme CYP3A4, resulting in the major active metabolite M19 (C4-pyrrolidine-hydroxylated rolapitant) and a number of inactive metabolites. Rolapitant is mainly excreted via the feces (52–89%) in unchanged form, and to a lesser extent via the urine (9–20%) in form of its inactive metabolites. Elimination half-life is about seven days (169 to 183 hours) over a wide dosing range.[8]
Chemistry
The drug is used in form of rolapitant hydrochloride monohydrate, a white to off-white, slightly hygroscopic crystalline powder. Its maximum solubility in aqueous solutions is at pH 2–4.[10]
Patents
WO 2003051840
PATENT
The preparation of diazaspirodecan-2-ones for example, 8-[{1-(3,5-Bis-(trifluoromethyl)phenyl)-ethoxy}-methyl]-8-phenyl-1,7-diaza-spiro[4.5]decan-2-one, for example, (5S,8S)-8-[{(1R)-1-(3,5-Bis-(trifluoromethyl)phenyl)-ethoxy}-methyl]-8-phenyl-1,7-diazaspiro[4.5]decan-2-one (the compound of Formula I) has been described in U.S. Pat. No. 7,049,320 (the ‘320 patent), issued May 23, 2006, the disclosure of which is incorporated herein in its entirety by reference.

The compounds described in the ‘320 patent are classified as tachykinin compounds, and are antagonists of neuropeptide neurokinin-1 receptors (herein, “NK-1” receptor antagonists). Other NK1 receptor antagonists and their synthesis have been described, for example, those described in Wu et al, Tetrahedron 56, 3043-3051 (2000); Rombouts et al, Tetrahedron Letters 42, 7397-7399 (2001); and Rogiers et al, Tetrahedron 57, 8971-8981 (2001) and in published international application no. WO05/100358, each of which are incorporated herein in their entirety by reference.
“NK-1” receptor antagonists have been shown to be useful therapeutic agents, for example, in the treatment of pain, inflammation, migraine, emesis (vomiting), and nociception. Among many compounds disclosed in the above-mentioned ‘320 patent are several novel diazaspirodecan-2-ones, including the compound of Formula I, which are useful in the treatment of nausea and emesis associated with chemotherapy treatments (Chemotherapy-induced nausea and emesis, CINE).
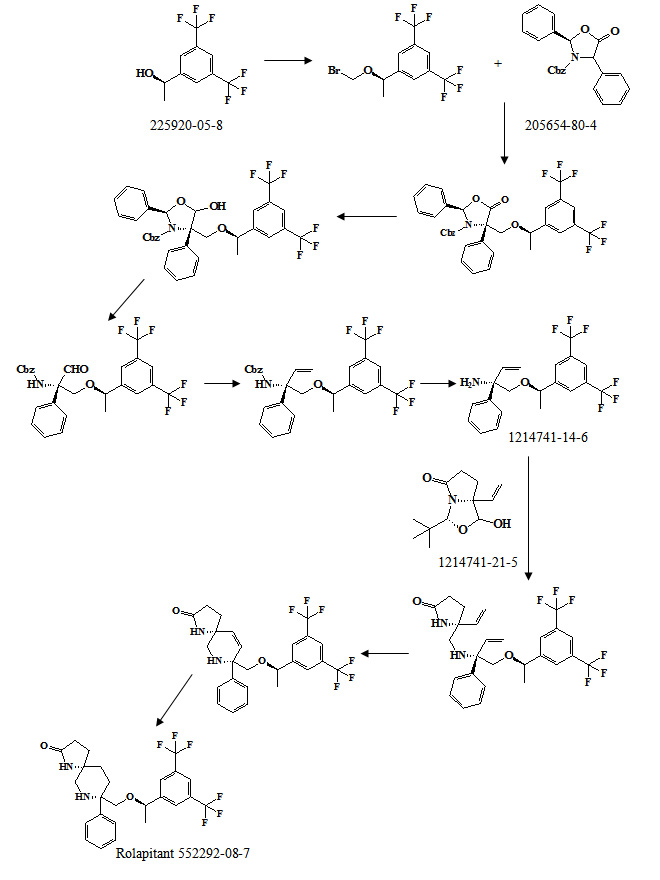
The synthesis method for preparing the compound of Formula I described in the ‘320 patent generally follows Scheme I in the provision of 8-[{1-(3,5-Bis-(trifluoromethyl)phenyl)-ethoxyl}-methyl]-8-phenyl-1,7-diaza-spiro[4.5]decan-2-one compounds.



The process for the preparation of the compound of Formula I described in the ‘320 patent is carried out in 18 individual steps from commercially available starting materials (see the ‘320 patent at col. 43, line 55 to col. 45, line 20; col. 75. line 55 to col. 80, line 21; col. 90 lines 35 to 63; and col. 98, line 1 to col. 99. line 24). In many steps of the process described in the ‘320 patent, intermediate compounds must be isolated or isolated and purified before use in a subsequent step, often utilizing column chromatography for this purpose.
PATENT
Examples 72a and 72b

Step 1:

To a solution of crude Compound 53 (19 g) in CH2Cl2 (300 ml) at RT, DIEA (15 ml, 0.087 mol) was added, followed by triphosgene (4.34 g, 0.015 mol). The mixture was stirred at RT for 18 h and was filtered through a pad of silica. Solvents were removed in vacuum to give crude Compound 60 as yellow oil which was used in the next reaction without further purifications.
Step 2:

To the crude Compound 60 in THF (200 ml) at 0° C., LiBH4 (1.26 g, 0.058 mol) was added in small portions. The mixture was stirred at RT for 18 h before quenching with saturated NH4Cl solution. Water and EtOAc were added to the mixture. Layers were separated and the aqueous layer was extracted with EtOAc (100×2). The combined organic layers were dried (MgSO4) and filtered. Solvents were removed in vacuum and purification by column chromatography [hexane-EtOAc, 4:1 (v/v)] gave Compound 61 (12.9 g, 62% overall) as white foam.
Step 3:
Oxalyl chloride (4.2 ml, 0.048 mol) was added to a solution of DMSO (6.8 m[, 0.096) in CH2Cl2 (300 ml) at −78° C. under N2. The mixture was stirred at −78° C. for 15 min before a solution of Compound 61 (8.5 g, 0.012 mol) in CH2Cl2 (100 ml) was added. The mixture was stirred at −78° C. for a further 1 h and Et3N (23.5 ml) was added. The cooling bath was removed and the mixture was warmed to RT before it was quenched with saturated NaHCO3 solution. Layers were separated and the aqueous was extracted with CH2Cl2 (150 ml×2). The combined organic layers were dried (MgSO4) and filtered. Removal of solvents in vacuum gave an aldehyde as yellow oil. To a mixture of NaH (1.44 g, 0.036 mol) in THF at 0° C., methyl diethylphosphonoacetate (6.6 ml, 0.036 mol) was added. The mixture was stirred at 0° C. for 15 min and a solution of aldehyde in THF (100 ml) was added. The cooling bath was removed and the mixture was stirred at RT for 1 h. The reaction was quenched with saturated NH4Cl solution. Water and EtOAc were added to the mixture. Layers were separated and the aqueous layer was extracted with EtOAc (200 ml×2). The combined organic layers were dried (MgSO4) and filtered. Solvents were removed in vacuum and purification by column chromatography [hexane-EtOAc, 4:1 (v/v)] gave an ester as white foam. The ester was dissolved in EtOH (100 ml) and a catalytic amount of palladium (1.28 g, 10% on carbon) was added. The mixture was shaken under H2 (50 psi) for 2 days. Catalytic amount of Pd(OH)2 (20% on carbon) was then added to the mixture and the mixture was again shaken under H2 (50 psi) for 5 h. The mixture was filtered through a pad of Celite and solvents were removed in vacuum to give a white foam. The foam was then dissolved in CH2Cl2 (200 ml) and TFA (8.9 ml, 0.12 mol) was added. The mixture was stirred at RT for 18 h and was cooled at 0° C. before it was neutralized with saturated NaHCO3 solution. Water and EtOAc were added to the mixture. Layers were separated and the aqueous layer was extracted with EtOAc (200 ml×2). The combined organic layers were dried (MgSO4) and filtered. Solvents were removed in vacuum to give a yellow oil. The oil was dissolved in CH3OH (50 ml) and a catalytic amount of K2CO3 (166 mg, 0.0012 mol) was added. The mixture was heated at 60° C. for 2 h. After being cooled to RT, the mixture was filtered through a pad of silica and solvents were removed in vacuum. Purification by column chromatography (EtOAc) gave the mixture of two isomers Example 72a and 72b (2.3 g, 38% overall) as white foam. Separation by HPLC using Chiralcel OD [hexane-isopropanol, 95:5 (v/v)] gave the less polar major isomer Example 72a as white foam. Electrospray MS [M+1]+=501.1. Continuous elution with the same solvent system gave the more polar minor isomer Example 72b as colorless oil.
Electrospray MS [M+1]+=501.1.





Example 6 Preparation of Formula I Compound Salt: (5S,8S)-8-({(1R)-1-[3,5-Bis(trifluoromethyl)phenyl]ethoxy}methyl)-8-phenyl-1,7-diazaspiro[4.5]decan-2-one hydrochloride monohydrate

…………………


https://www.google.it/patents/US8552191?hl=it&dq=WO+2008118328&ei=alDCUs-_KYiIrQeg3oCwDw&cl=en
……………
update added
(RTTNews.com) – TESARO Inc. ( TSRO ) announced positive top-line results from the third and final Phase 3 trial of rolapitant, an investigational neurokinin-1 or NK-1 receptor antagonist in development for the prevention of chemotherapy-induced nausea and vomiting (CINV).
The rolapitant arm in this trial, which enrolled patients receiving cisplatin-based, highly emetogenic chemotherapy or HEC, successfully achieved statistical significance over the standard therapy arm for the primary and all secondary endpoints. The adverse event profile for rolapitant remains consistent with that seen in previous clinical studies.
The third Phase 3 study of rolapitant was an international, multicenter, randomized, double-blind, active-controlled study that enrolled 532 cancer patients receiving cisplatin-based chemotherapy regimens at a dose equal to or greater than 60 mg/m2. Patients were randomized to receive either control, which consisted of a 5-HT3 receptor antagonist plus dexamethasone, or 200 milligrams of oral rolapitant plus control. The rolapitant arm in this study successfully achieved statistical significance over the control arm for the primary endpoint of complete response (CR) in the delayed phase of CINV.
In addition, the rolapitant arm also successfully achieved statistical significance over the control arm for the key secondary endpoints of CR in the acute (0 to 24 hour) and overall (0 to 120 hour) phases of CINV, for the secondary endpoint of no significant nausea, and for all other secondary endpoints.
Safety and tolerability data for patients who received rolapitant were similar to the results for those who received control, and were consistent with earlier clinical studies. The most frequently observed adverse events were balanced across treatment arms and included fatigue, constipation and loss of appetite.
The company noted that preparations continue in support of a submission of a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) in mid-2014.
The oral rolapitant NDA will include data from one Phase 3 study in patients receiving moderately emetogenic chemotherapy (MEC), in addition to one Phase 2 and two Phase 3 trials in patients receiving cisplatin-based, highly emetogenic chemotherapy (HEC), including the trial announced today.
The top-line results of the Phase 3 trial in MEC and the prior Phase 3 trial in HEC were previously announced by TESARO in December 2013.
Rolapitant is an investigational agent and, as such, has not been approved by the U.S. FDA or any regulatory agencies.
CLIP
Rolapitant Hydrochloride Hydrate (Varubi)



-
67 . Syed, Y. Y. Rolapitant: First Global Approval Drugs 2015, 75, 1941– 1945 DOI: 10.1007/s40265-015-0485-8
-
68.Duffy, R. A.; Morgan, C.; Naylor, R.; Higgins, G. A.; Varty, G. B.; Lachowicz, J. E.; Parker, E. M. Rolapitant (SCH 619734): A Potent, Selective and Orally Active Neurokinin NK1 Receptor Antagonist with Centrally-mediated Antiemetic Effects in Ferrets Pharmacol., Biochem. Behav. 2012, 102, 95– 100 DOI: 10.1016/j.pbb.2012.03.021
-
69.Janelsins, M. C.; Tejani, M. A.; Kamen, C.; Peoples, A. R.; Mustian, K. M.; Morrow, G. R. Current Pharmacotherapy for Chemotherapy-induced Nausea and Vomiting in Cancer Patients Expert Opin. Pharmacother. 2013, 14, 757– 766 DOI: 10.1517/14656566.2013.776541
-
70.Navari, R. M. Rolapitant for the Treatment of Chemotherapy-induced Nausea and Vomiting Expert Rev. Anticancer Ther. 2015, 15, 1127– 1133 DOI: 10.1586/14737140.2015.1088787
-
71.Romero, D. Chemotherapy Rolapitant – a New and Safer Antiemetic Agent Nat. Rev. Clin. Oncol. 2015, 12,562 DOI: 10.1038/nrclinonc.2015.144
-
72.(a) Schwartzberg, L. S.; Modiano, M. R.; Rapoport, B. L.; Chasen, M. R.; Gridelli, C.; Urban, L.; Poma, A.;Arora, S.; Navari, R. M.; Schnadig, I. D. Safety and Efficacy of Rolapitant for Prevention of Chemotherapy-induced Nausea and Vomiting after Administration of Moderately Emetogenic Chemotherapy or Anthracycline and Cyclophosphamide Regimens in Patients with Cancer: a Randomised, Active-controlled, Double-blind, Phase 3 Trial Lancet Oncol. 2015, 16, 1071– 1078 DOI: 10.1016/S1470-2045(15)00034-0
(b) Rapoport, B.; Schwartzberg, L.; Chasen, M.; Powers, D.; Arora, S.;Navari, R.; Schnadig, I. Efficacy and Safety of Rolapitant for Prevention of Chemotherapy-induced Nausea and Vomiting Over Multiple Cycles of Moderately or Highly Emetogenic Chemotherapy Eur. J. Cancer 2016,57, 23– 30 DOI: 10.1016/j.ejca.2015.12.023
-
73.Wu, G. G.; Werne, G.; Fu, X.; Orr, R. K.; Chen, F. X.; Cui, J.; Sprague, V. M.; Zhang, F.; Xie, J.; Zeng, L.;Castellanos, L. P.; Chen, Y.; Poirier, M.; Mergelsberg, I. Process and Intermediates for the Synthesis of 8-[[1-[3,5-bis-(trifluoromethyl)phenyl]ethoxy]methyl]-8-phenyl-1,7-diazaspiro[4.5]decan-2-one Compounds. WO 2010028232A1, 2010.
-
74.Dikshit, D. K.; Maheshwari, A.; Panday, S. K. Self Reproduction of Chirality in Pyroglutamates: Reactions at α-Position with Electrophiles Tetrahedron Lett. 1995, 36, 6131– 6134 DOI: 10.1016/0040-4039(95)01160-J
-
75.O’Donnell, M. J.; Fang, Z.; Ma, X.; Huffman, J. C. New Methodology for the Synthesis of α,α-Dialkylamino Acids Using the ″Self-regeneration of Stereocenters″ Method: α-Ethyl-α-phenylglycine Heterocycles 1997,46, 617– 630 DOI: 10.3987/COM-97-S83
-
76.Paliwal, S.; Reichard, G. A.; Wang, C.; Xiao, D.; Tsui, H.-C.; Shih, N.-Y.; Arredondo, J. D.; Wrobleski, M. L.;Palani, A. Preparation of Pyrrolidine and Piperidine Derivatives for Therapeutic Use as Neurokinin 1 (NK1) Receptor Antagonists. WO 2003051840A1, 2003.
REF
HETEROCYCLES 1997 46 PG 617 630
Paper | Special issue | Vol 46, No. 1, 1997, pp.617-630
Published online, 1st January, 1970
■ New Methodology for the Synthesis of α,α-Dialkylamino Acids Using the “Self-Regeneration of Stereocenters” Method: α-Ethyl-α-phenylglycine
Martin J. O’Donnell,* Zhiqiang Fang, Xiaojun Ma, and John C. Huffman
*Department of Chemistry, Indiana University-Purdue University at Indianapolis, Indianapolis, IN 46202, U.S.A.
Abstract
The stereoselective room temperature ethylations of protected oxazolidinones from phenylglycine by phase-transfer catalysis or with KOtBu as base are used to prepare optically active α-ethyl-α-phenylglycine.
PATENT
https://patents.google.com/patent/CN106866669A/en
⑴ Route A:

[0005] ⑵ Route B:

[0007] (3) Route C:

[0009] Scheme C, wherein the method further comprises synthesizing Via, namely:

Won] now, with respect to the other two routes, from the reaction step, time costs, material costs, product yield and product purity of view, comparing the current line C is respected, it is more suitable for production. But even so, there are still a number of route C the following questions:
[0012] [1], the synthesis of compound V, there is a slow reaction, and the reaction was not complete and so on;
[0013] [2], when Via a salt, the desired product is low chiral purity and yield to be improved;
[0014] [3], when VIII recrystallized grain size to be improved.
CLIP
| References |
1: Gan TJ, Gu J, Singla N, Chung F, Pearman MH, Bergese SD, Habib AS, Candiotti KA, Mo Y, Huyck S, Creed MR, Cantillon M; Rolapitant Investigation Group. Rolapitant for the prevention of postoperative nausea and vomiting: a prospective, double-blinded, placebo-controlled randomized trial. Anesth Analg.
2011 Apr;112(4):804-12. Epub 2011 Mar 8. PubMed PMID: 21385988.
2. Reddy GK, Gralla RJ, Hesketh PJ. Novel neurokinin-1 antagonists as antiemetics for the treatment of chemotherapy-induced emesis. Support Cancer Ther. 2006 Apr 1;3(3):140-2. PubMed PMID: 18632487.
3. Drug Data Rep 2003, 25(8): 703
4. A multicenter, randomized, double blind, active-controlled study of the safety and efficacy of rolapitant for the prevention of chemotherapy-induced nausea and vomiting (CINV) in subjects receiving moderately emetogenic chemotherapy (NCT01500226)
ClinicalTrials.gov Web Site 2012, February 06
5. Efficacy and safety of rolapitant, a novel NK-1 receptor antagonist, for the prevention of chemotherapy-induced nausea and vomiting in subjects receiving highly emetogenic chemotherapy
48th Annu Meet Am Soc Clin Oncol (ASCO) (June 1-5, Chicago) 2012, Abst 9077
6. Proposed international nonproprietary names (Prop. INN): List 97
WHO Drug Inf 2007, 21(2): 160
References
- ^ Jump up to:a b “Varubi (rolapitant) Tablets, for Oral Use. Full Prescribing Information” (PDF). TESARO, Inc. 1000 Winter St., #3300, Waltham, MA 02451.
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names (Rec. INN): List 59” (PDF). World Health Organization. p. 64. Retrieved 5 October 2016.
- ^ Duffy, R. A; Morgan, C; Naylor, R; Higgins, G. A; Varty, G. B; Lachowicz, J. E; Parker, E. M (2012). “Rolapitant (SCH 619734): a potent, selective and orally active neurokinin NK1 receptor antagonist with centrally-mediated antiemetic effects in ferrets”. Pharmacol Biochem Behav. 102 (1): 95–100. doi:10.1016/j.pbb.2012.03.021. PMID 22497992.
- ^ Jordan, K; Jahn, F; Aapro, M (2015). “Recent developments in the prevention of chemotherapy-induced nausea and vomiting (CINV): a comprehensive review”. Ann Oncol. 26 (6): 1081–90. doi:10.1093/annonc/mdv138. PMID 25755107.
- ^ Nasir, S. S; Schwartzberg, L. S (2016). “Recent Advances in Preventing Chemotherapy-Induced Nausea and Vomiting”. Oncology. 30 (8): 750–62. PMID 27539626.
- ^ Rapoport, B; Schwartzberg, L; Chasen, M; Powers, D; Arora, S; Navari, R; Schnadig, I (2016). “Efficacy and safety of rolapitant for prevention of chemotherapy-induced nausea and vomiting over multiple cycles of moderately or highly emetogenic chemotherapy”. Eur J Cancer. 57: 23–30. doi:10.1016/j.ejca.2015.12.023. PMID 26851398.
- ^ Chasen, M. R; Rapoport, B. L (2016). “Rolapitant for the treatment of chemotherapy-induced nausea and vomiting: a review of the clinical evidence”. Future Oncol. 12 (6): 763–78. doi:10.2217/fon.16.11. PMID 26842387.
- ^ Jump up to:a b c d e f g h “Varuby: EPAR – Product Information” (PDF). European Medicines Agency. 2017-05-31.
- ^ Jump up to:a b FDA Professional Drug Information on Varubi. Accessed 2017-10-11.
- ^ Jump up to:a b “Varuby: EPAR – Public assessment report” (PDF). European Medicines Agency. 2017-05-31.
 |
|
| Clinical data | |
|---|---|
| Pronunciation | /roʊˈlæpɪtænt/ roh-LAP-i-tant |
| Trade names | Varubi (US), Varuby (EU) |
| Synonyms | SCH 619734 |
| AHFS/Drugs.com | varubi |
| License data | |
| Routes of administration |
By mouth (tablets) |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | nearly 100% |
| Protein binding | 99.8% |
| Metabolism | CYP3A4 |
| Metabolites | C4-pyrrolidine-hydroxylated rolapitant (major) |
| Elimination half-life | 169–183 hours |
| Excretion | Feces (52–89%), urine (9–20%)[1] |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| Chemical and physical data | |
| Formula | C25H26F6N2O2 |
| Molar mass | 500.476 g/mol |
| 3D model (JSmol) | |