














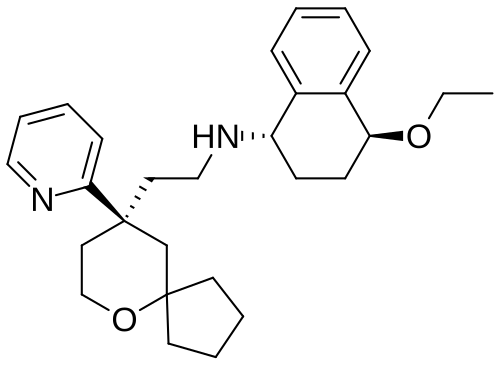
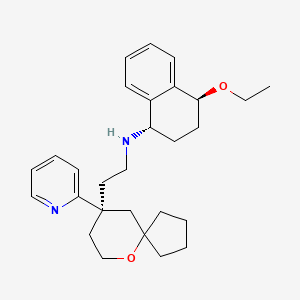
Tegileridine
- YFJS8L4TGU
- CAS 2095345-66-5
- (9R)-N-((1S,4S)-4-Ethoxy-1,2,3,4-tetrahydro-1-naphthalenyl)-9-(2-pyridinyl)-6-oxaspiro(4.5)decane-9-ethanamine
- 434.6 g/mol
WeightAverage: 434.624
Monoisotopic: 434.293328472
Chemical FormulaC28H38N2O2
(1S,4S)-4-ethoxy-N-[2-[(9R)-9-pyridin-2-yl-6-oxaspiro[4.5]decan-9-yl]ethyl]-1,2,3,4-tetrahydronaphthalen-1-amine
- (9R)-N-((1S,4S)-4-Ethoxy-1,2,3,4-tetrahydro-1-naphthalenyl)-9-(2-pyridinyl)-6-oxaspiro(4.5)decane-9-ethanamine
- (1S,4S)-4-Ethoxy-N-(2-((R)-9-(pyridin-2-yl)-6-oxaspiro(4.5)decan-9-yl)ethyl)-1,2,3,4-tetrahydronaphthalen-1-amine
- (1S,4S)-4-Ethoxy-N-(2-((R)-9-(pyridin-2-yl)-6-oxaspiro(4.5)decane-9-yl)ethyl)-1,2,3,4-tetrahydronaphthalen-1-amine
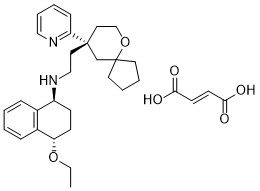
Tegileridine fumarate
CAS#2245827-85-2 (fumarate)
Chemical Formula: C32H42N2O6
Exact Mass: 550.3000
Molecular Weight: 550.70
CHINA 2025, APPROVALS 2025, AISUTE, Jiangsu Hengrui
Tegileridine is under investigation in clinical trial NCT06458400 (To Evaluate the Efficacy and Safety of Tegileridine and Oliceridine Injections in the Treatment of Postoperative Pain).
Tegileridine is a drug which acts as a μ-opioid receptor agonist. It is closely related to compounds such as oliceridine, TRV734, and SHR9352, and shares a similar profile as a biased agonist selective for activation of the G-protein signalling pathway over β-arrestin 2 recruitment.[1]
In January 2024, tegileridine was approved in China for the treatment of moderate to severe pain after abdominal surgery.[2]
SYN
CN107001347
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN203399246&_cid=P20-METU4Y-21400-1
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017063509&_cid=P20-METU6J-22458-1
[0184](S)-1-Ethyl-N-(2-((R)-9-(pyridin-2-yl)-6-oxaspiro[4.5]decan-9-yl)ethyl)-1,2,3,4-tetrahydroquinolin-1-amine 1
[0185](R)-1-ethyl-N-(2-((R)-9-(pyridin-2-yl)-6-oxaspiro[4.5]decan-9-yl)ethyl)-1,2,3,4-tetrahydroquinolin-1-amine 2
[0186]
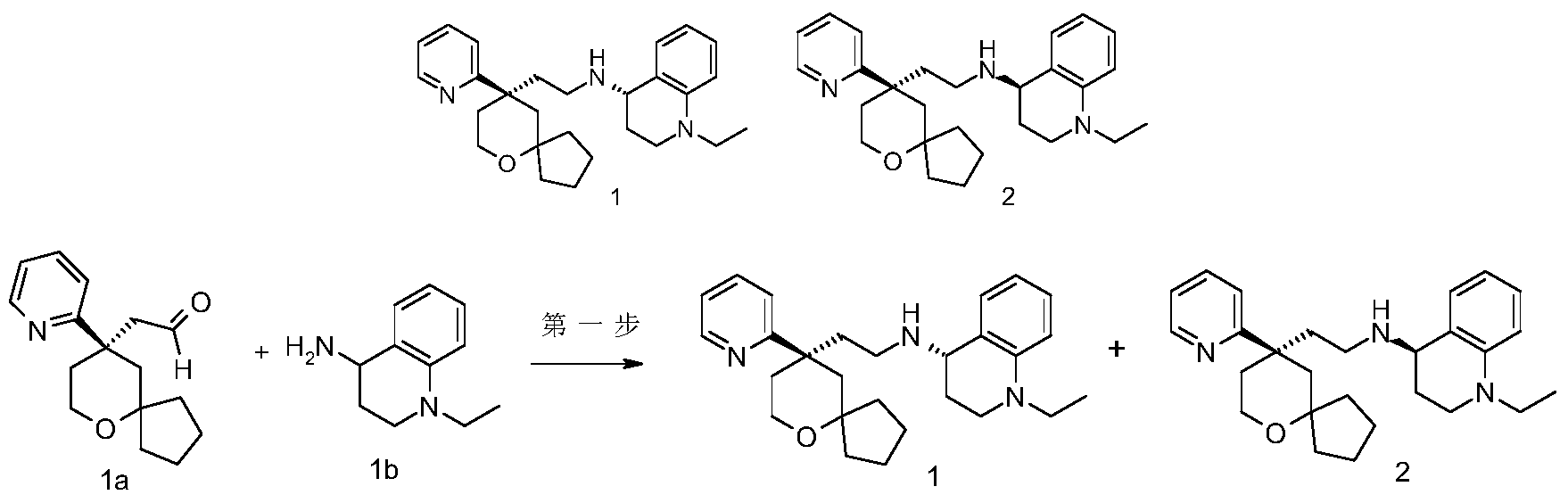
[0187](R)-2-(9-(pyridin-2-yl)-6-oxaspiro[4,5]decane-9-yl)acetaldehyde 1a (294 mg, 1.135 mmol, prepared by the method disclosed in patent application “WO2012129495”) and 1-ethyl-1,2,3,4-tetrahydroquinolin-4-amine 1b (200 mg, 1.135 mmol, prepared by the method disclosed in patent application “WO2014078454”) were dissolved in 15 mL of dichloromethane, stirred for 1 hour, and sodium triacetoxyborohydride (1.203 g, 5.675 mmol) was added and stirred for 16 hours. 20 mL of water was added, and the mixture was extracted with dichloromethane (20 mL×3). The organic phases were combined, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by high performance liquid chromatography to obtain the title product, 1-ethyl-N-(2-((R)-9-(pyridin-2-yl)-6-oxaspiro[4.5]decan-9-yl)ethyl)-1,2,3,4-tetrahydroquinolin-1-amine. Chiral preparation was performed (separation conditions: chiral preparative column Superchiral S-AD (Chiralway), 2 cm ID*25 cm, 5 um; mobile phase: CO
2 :methanol:diethanolamine=75:25:0.05, flow rate: 50 g/min). The corresponding fractions were collected and concentrated under reduced pressure to give the title products 1 (98 mg, brown oil) and 2 (95 mg, yellow solid).
[0190]Chiral HPLC analysis: retention time 4.028 minutes, chiral purity: 99.7% (chromatographic column: Superchiral S-AD (Chiralway), 0.46 cm ID*15 cm, 5 μm; mobile phase: CO2: methanol: diethanolamine = 75:25:0.05 (v/v/v))
[0191]
1H NMR(400MHz,DMSO-d 6)δ8.54(s,1H),7.72(s,1H),7.45(d,1H),7.20(s,1H),6.95(s,1H),6.78(d,1H),6.52(d,1H),6.37(s,1H),3.60(br,2H),3.18-3.43(m,3H),2.99(m,1H),2.33-2.45(m,3H),1.77-1.99(m,3H),1.19-1.60(m,12H),1.00-1.06(m,4H),0.63(m,1H).
[0194]Chiral HPLC analysis: retention time 3.725 minutes, chiral purity: 99.8% (chromatographic column: Superchiral S-AD (Chiralway), 0.46 cm ID*15 cm, 5 μm; mobile phase: CO2: methanol: diethanolamine = 75:25:0.05 (v/v/v))
[0195]
1H NMR(400MHz,DMSO-d 6)δ8.53(s,1H),7.72(s,1H),7.46(d,1H),7.20(s,1H),6.97(s,1H),6.85(d,1H),6.54(d,1H),6.40(s,1H),3.61(br,2H),3.17-3.25(m,3H),3.00-3.01(m,1H),2.33-2.46(m,3H),1.78-1.97(m,3H),1.24-1.65(m,12H),1.01-1.06(m,4H),0.61(m,1H).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US306969245&_cid=P20-METUA8-25189-1
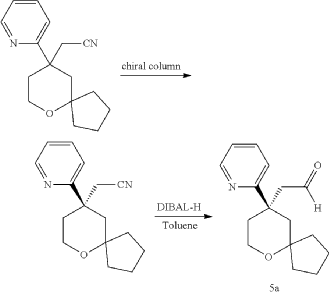
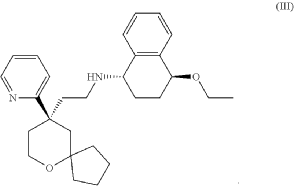
Embodiment 1: Preparation of (1S,4S)-4-ethoxy-N-(2-((R)-9-(pyridin-2-yl)-6-oxaspiro[4.5]decan-9-yl)ethyl)-1,2,3,4-tetrahydronaphthalen-1-amine
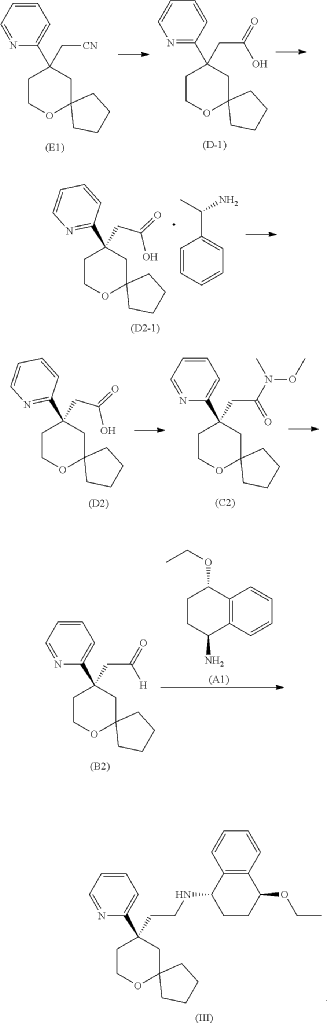
Step One: Synthesis of Intermediate (D-1)
Step Two: Synthesis of Intermediate (D2-1)
Step Three: Synthesis of Intermediate (D2)
Step Four: Synthesis of Intermediate (C2)
Step Five: Synthesis of Intermediate (B2)
Step Six: Synthesis of the Compound Represented by Formula (III)
SYN
SYN
Tegileridine fumarate, developed by Jiangsu Hengrui Pharmaceuti
cals Co., Ltd., is a novel small-molecule analgesic that functions as a
complete opioid receptor agonist with relative selectivity for -opioid
receptors (MOR). It is marketed under the brand name Aisute. In 2024,
the NMPA approved Tegileridine fumarate injection for the treatment of moderate to severe pain following abdominal surgery. Tegileridine ex
erts its analgesic effects by activating MOR, leading to inhibition of
adenylate cyclase activity, decreased intracellular cAMP levels, and
subsequent modulation of ion channel conductance. This results in hy
perpolarization of neuronal membranes and reduced neuronal excit
ability, effectively alleviating pain. The clinical efficacy of Tegileridine
was evaluated in a Phase III randomized, double-blind, placebo-
controlled trial (NCT05012516) involving patients experiencing mod
erate to severe pain after abdominal surgery. The research indicated that
Tegileridine offered substantial alleviation of pain in contrast to the
placebo. It manifested a quick-acting property, and its analgesic effects
endured throughout the period of observation. In terms of toxicity,
Tegileridine was typically well-tolerated by the subjects. The most
frequently encountered adverse reactions were nausea, vomiting, and
dizziness, all of which were of mild to moderate intensity. Importantly,
Tegileridine exhibited a favorable safety profile with a lower incidence
of gastrointestinal adverse reactions compared to traditional MOR ag
onists, potentially offering an improved therapeutic window for post
operative pain management. The approval of Tegileridine provides a
new treatment option for patients suffering from moderate to severe
postoperative pain, particularly following abdominal surgeries,
addressing a significant clinical need in pain management [72,73].
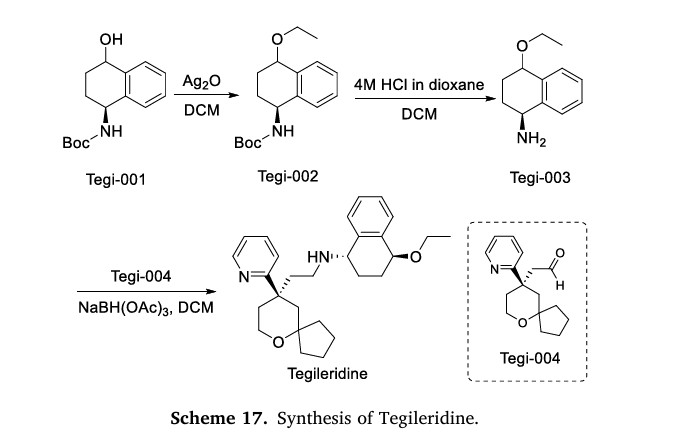
The synthesis of Tegileridine fumarate, illustrated in Scheme 17,
begins with nucleophilic substitution reaction involving Tegi-001 to
yield Tegi-002 [74]. Tegi-002 is subsequently acidified to produce
Tegi-003. Finally, Tegi-003 undergoes reductive amination with
Tegi-004 to synthesize Tegileridine.
[72] S. Dhillon, Correction: tegileridine: first approval, Drugs 84 (2024) 1011.
[73] S. Dhillon, Tegileridine: first approval, Drugs 84 (2024) 717–720.
[74] X. Li, B. Feng, Y. Chen, T. Liu, F. He, M. He, W. Tao, P. Sun, Oxa Spiro Derivative
Useful in Treatment of Pain and pain-related Disease and Its Preparation, 2017.
CN107001347A.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- WO 2017/063509, “Oxa spiro derivative, preparation method therefor, and applications thereof in medicines”, published 10 April 2018, assigned to Jiangsu Hengrui Medicine Company and Shanghai Hengrui Pharmaceutical Company Ltd .
- Dhillon S (June 2024). “Tegileridine: First Approval”. Drugs. 84 (6): 717–720. doi:10.1007/s40265-024-02033-4. PMID 38771484.
| Clinical data | |
|---|---|
| Trade names | 艾苏特 |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2095345-66-5 |
| PubChem CID | 129049969 |
| UNII | YFJS8L4TGU |
| Chemical and physical data | |
| Formula | C28H38N2O2 |
| Molar mass | 434.624 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
{kind=link}
- New drugs approved by the NMPA in 2024: Synthesis and clinical applicationsPublication Name: European Journal of Medicinal ChemistryPublication Date: 2025-07-05PMID: 40262297DOI: 10.1016/j.ejmech.2025.117643
- Tegileridine: First ApprovalPublication Name: DrugsPublication Date: 2024-05-21PMID: 38771484DOI: 10.1007/s40265-024-02033-4
- Study of the mass balance, biotransformation and safety of [14C]SHR8554, a novel μ-opioid receptor injection, in healthy Chinese subjectsPublication Name: Frontiers in PharmacologyPublication Date: 2023-09-14PMCID: PMC10538116PMID: 37781692DOI: 10.3389/fphar.2023.1231102
- Oxa spiro derivative, preparation method therefor, and applications thereof in medicinesPublication Number: US-2018297988-A1Priority Date: 2015-10-15
- Oxa spiro derivative, preparation method therefor, and applications thereof in medicinesPublication Number: WO-2017063509-A1Priority Date: 2015-10-15
- Oxaspiro derivatives, methods of their manufacture, and their application in pharmaceuticalsPublication Number: JP-6824502-B2Priority Date: 2015-10-15Grant Date: 2021-02-03
- Oxa spiro derivatives, their preparation, and their applications in medicinePublication Number: KR-102703513-B1Priority Date: 2015-10-15Grant Date: 2024-09-06
- Opioid Receptor (MOR) Agonist Salt, Its Fumarate Salt I Crystalline Form, and Process for Making SamePublication Number: JP-7153030-B6Priority Date: 2017-04-14Grant Date: 2023-07-24
- Oxa spiro derivative, preparation method therefor, and applications thereof in medicinesPublication Number: EP-3354649-A1Priority Date: 2015-10-15
- Oxa spiro derivative, preparation method therefor, and applications thereof in medicinesPublication Number: EP-3354649-B1Priority Date: 2015-10-15Grant Date: 2019-12-04
- Oxaspiro derivative, process for its production and its application in medicinePublication Number: JP-2018534257-APriority Date: 2015-10-15
- Oxa spiro derivative, preparation method therefor, and applications thereof in medicinesPublication Number: US-10442793-B2Priority Date: 2015-10-15Grant Date: 2019-10-15
////////////Tegileridine, CHINA 2025, APPROVALS 2025, AISUTE, Jiangsu Hengrui, YFJS8L4TGU, 2095345-66-5, Tegileridine FUMARATE