The synthesis of peptides by amide bond formation between suitably protected amino acids is a fundamental part of the drug discovery process. However, the required coupling and deprotection reactions are routinely carried out in dichloromethane and DMF, both of which have serious toxicity concerns and generate waste solvent which constitutes the vast majority of the waste generated during peptide synthesis. In this work, propylene carbonate has been shown to be a green polar aprotic solvent which can be used to replace dichloromethane and DMF in both solution- and solid-phase peptide synthesis. Solution-phase chemistry was carried out with Boc/benzyl protecting groups to the tetrapeptide stage, no epimerisation occurred during these syntheses and chemical yields for both coupling and deprotection reactions in propylene carbonate were at least comparable to those obtained in conventional solvents. Solid-phase peptide synthesis was carried out using Fmoc protected amino acids on a ChemMatrix resin and was used to prepare the biologically relevant nonapeptide bradykinin with comparable purity to a sample prepared in DMF.

Boc-Ala-Phe-OBn 5a ref S1
Boc-Ala-OH (324 mg, 1.71 mmol) and HCl.H-Phe-OBn (500 mg, 1.71 mmol) were coupled according to the general coupling procedure. The residue was purified using flash column chromatography (35:65, EtOAc:PE) to give Boc-Ala-Phe-OBn 5a as a white crystalline solid (682 mg, 93%). RF = 0.34 (40:60, EtOAc:PE);
mp 95.6-96.3 °C;
[α]D 23 -27.7 (c 1.0 in MeOH);
IR (Neat) νmax 3347 (m), 3063 (w), 3029 (w), 2928 (m), 2852 (w), 1735 (w), 1684 (w) 1666 (w) and 1521 (s) cm-1;
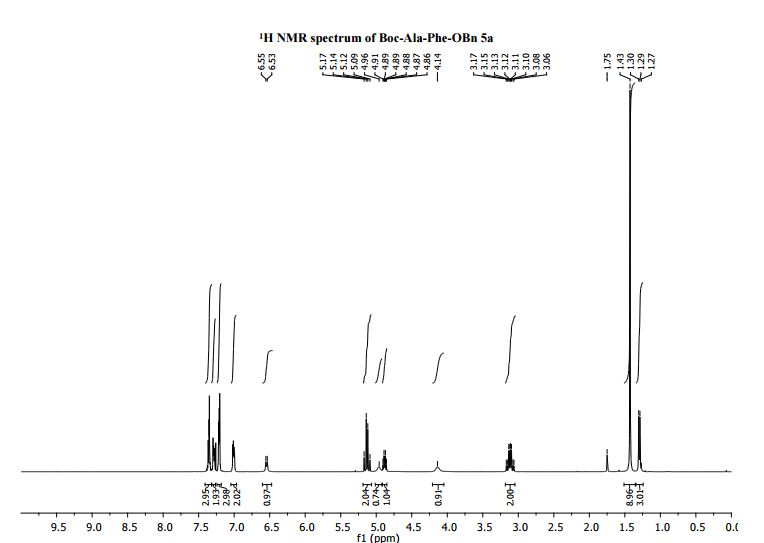
1H NMR (400 MHz, CDCl3): δ = 7.36-7.31 (m, 3H, ArH), 7.29-7.24 (m, 2H, ArH), 7.26-7.21 (m, 3H, ArH), 7.04-6.97 (m, 2H, ArH), 6.72 (d J 7.7 Hz, 1H, Phe-NH), 5.16-5.10 (m, 1H, Ala-NH), 5.13 (d J 12.1 Hz, 1H, OCH2Ph), 5.07 (d J 12.1 Hz, 1H, OCH2Ph), 4.88 (dt, J 7.7, 5.9 1H, PheNCH), 4.11 (br, 1H, Ala-NCH), 3.13 (dd J 13.9, 6.1 Hz, 1H, CH2Ph), 3.08 (dd J 13.9, 6.1 Hz, 1H, CH2Ph), 1.41 (s, 9H, C(CH3)3), 1.29 (d J 6.6 Hz, 3H, CH3);
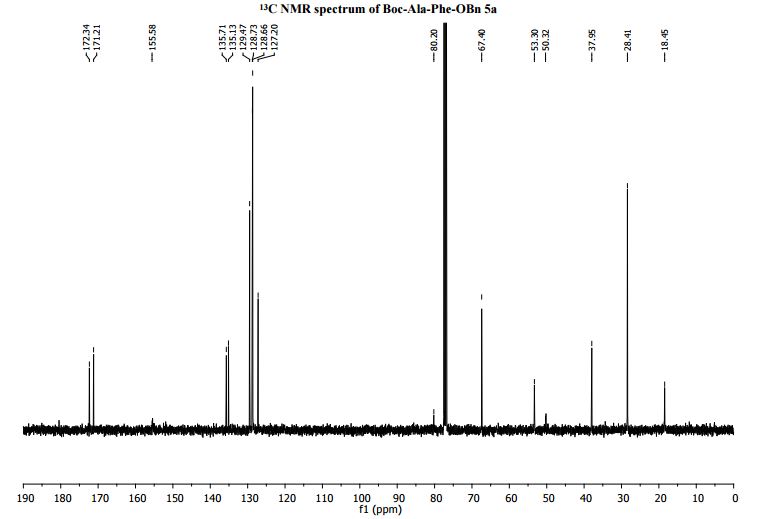
13C NMR (100 MHz, CDCl3): δ = 172.3 (C=O), 171.2 (C=O), 155.6 (NC=O), 135.7 (ArC), 135.1 (ArC), 129.5 (ArCH), 128.7 (ArCH), 128.6 (ArCH), 127.2 (ArCH), 80.2 (CMe3), 67.4 (OCH2Ph), 53.3 (Phe-NCH), 50.3 (Ala-NCH), 38.0 (CH2Ph), 28.4 (C(CH3)3), 18.5 (CH3);
MS (ESI) m/z 449 [(M+Na)+ , 100]; HRMS (ESI) m/z calculated for C24H30N2O5Na 449.2048 (M+Na)+ , found 449.2047 (0.6 ppm error).
S1 J. Nam, D. Shin, Y. Rew and D. L. Boger, J. Am. Chem. Soc., 2007, 129, 8747–8755; Q. Wang, Y. Wang and M. Kurosu, Org. Lett., 2012, 14, 3372–3375.
General procedure for peptide coupling reactions in PC To a suspension of an N-Boc-amino acid (1.0 eq.) and an amino acid or peptide benzyl ester (1.0 eq.) in PC (5 mL mmol-1), at 0 °C, was added a solution of HOBt (1.1 eq.) and i Pr2EtN (3.0 eq.) in a minimal quantity of PC. EDC (1.1 eq.) was added dropwise and the reaction mixture was allowed to stir at room temperature for 16h. The reaction mixture was then diluted using EtOAc (50 mL) and washed with 1M HClaq (3 × 25 mL), saturated Na2CO3 (3 × 25 mL) and H2O (3 × 25 mL). The organic layer was dried (MgSO4 ), filtered and concentrated in vacuo. Any residual PC was removed via short path distillation. Purification details for each peptide and characterising data are given in the supplementary information. General procedure for Boc deprotections in PC An N-Boc-peptide benzyl ester (1.0 eq.) was dissolved in a minimum amount of PC and trifluoroacetic acid (60 eq.) was added. The reaction mixture was allowed to stir for 3h. at room temperature before being concentrated in vacuo. Any residual PC was removed via short path distillation. Characterising data for each deprotected peptide are given in the supplementary information.
Procedure for Boc deprotection of dipeptide 5a using HCl in PC Boc-Ala-Phe-OBn 5a (50 mg, 0.117 mmol) was dissolved in PC (2.34 mL). MeOH (0.40 mL, 9.8 mmol) was added and the solution cooled to 0 o C. Acetyl chloride (0.67 mL, 9.36 mmol) was added dropwise and the solution allowed to stir at room temperature for 2h. Then, PC was removed by short path distillation. The residue was suspended in Et2O and stirred for 5 minutes before being filtered to give HCl.Ala-Ph-OBn as a white solid (32.4 mg, 76%).
Propylene carbonate 1 has been shown to be a green replacement for reprotoxic amide based solvents which are widely used in peptide synthesis. Both solution- and solidphase peptide synthesis can be carried out in propylene carbonate using acid and base labile amine protecting groups respectively. No significant racemisation of the activated amino acids occurs in propylene carbonate and the viability of solid-phase peptide synthesis in propylene carbonate was demonstrated by the synthesis of the nonapeptide bradykinin.