














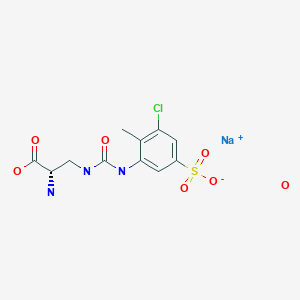
Upacicalcet sodium hydrate, ウパシカルセトナトリウム水和物
CAS 2052969-18-1
1333218-50-0 free
PMDA JAPAN APPROVED 2021/6/23, Upasita
Calcium sensing receptor agonist
(2S)-2-amino-3-[(3-chloro-2-methyl-5-sulfophenyl)carbamoylamino]propanoic acid
| Formula |
C11H13ClN3O6S. Na. xH2O
|
|---|
- OriginatorAjinomoto Pharma
- DeveloperSanwa Kagaku Kenkyusho
- ClassAmines; Chlorobenzenes; Propionic acids; Small molecules; Sulfonic acids; Toluenes
- Mechanism of ActionCalcium-sensing receptor agonists
- RegisteredSecondary hyperparathyroidism
- 25 Jun 2021Chemical structure information added
- 23 Jun 2021Sanwa Kagaku Kenkyusho and Kissei Pharmaceutical agree to co-promote upacicalcet in Japan for Secondary hyperparathyroidism
- 23 Jun 2021Registered for Secondary hyperparathyroidism in Japan (IV) – First global approval
| Upacicalcet Sodium Hydrate
Monosodium 3-({[(2S)-2-amino-2-carboxyethyl]carbamoyl}amino)-5-chloro-4-methylbenzenesulfonate hydrate C11H13ClN3NaO6S▪xH2O |
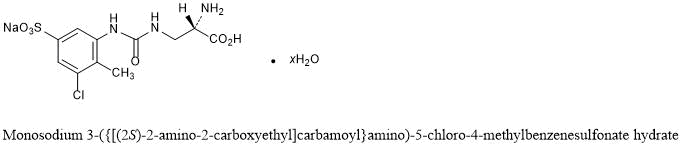
Announcement of Marketing Authorization Approval in Japan and Co-promotion Agreement of UPASITA® IV Injection Syringe for the Treatment of Secondary Hyperparathyroidism in Dialysis Patients
SANWA KAGAKU KENKYUSHO Co., Ltd. (Head Office: Nagoya, President and CEO : Shusaku Isono, Suzuken Group, ; “SANWA KAGAKU”) has received Marketing Authorization approval today for UPASITA® IV Injection Syringes (generic name: Upacicalcet Sodium Hydrate; “UPASITA®”) for the treatment of secondary hyperparathyroidism in patients on hemodialysis.
UPASITA® was created by Ajinomoto Pharmaceuticals Co., Ltd. (currently EA Phama Co., Ltd.) and developed by SANWA KAGAKU for the treatment of secondary hyperparathyroidism under a licensing agreement with EA Pharma. UPASITA® acts on calcium sensing receptor in the parathyroid and suppresses excessive secretions of parathyroid hormones (PTH). UPASITA® is administered by intravenous injection to dialysis patients through dialysis circuit by physicians or medical staffs upon completion of dialysis and such administration is expected to reduce the burden of patients with many oral medications whose drinking water volume is severely restricted.
Regarding provision of medical and drug information, SANWA KAGAKU entered into a co-promotion agreement in Japan with Kissei Pharmaceutical Co., Ltd. (Head Office: Matsumoto, Nagano; Chairman and CEO: Mutsuo Kanzawa ; “Kissei”). SANWA KAGAKU will handle the production, marketing, and distribution of the Product while SANWA KAGAKU and Kissei collaboratively promote it to medical institutions in the field in accordance with the agreement. Through the co-promotion activity in the field, SANWA KAGAKU and Kissei will contribute to the treatment of dialysis patients suffering from secondary hyperparathyroidism.
《Reference》
About secondary hyperparathyroidism (SHPT)
SHTP is one of complications that occur as chronic kidney disease (chronic kidney failure) progresses and is a pathological condition where excessive PTH is secreted by the parathyroid gland. It has been reported that excessive secretion of parathyroid hormone promotes efflux of phosphorus and calcium from the bone into the blood, thereby increasing the risk of developing bone fractures and arteriosclerosis due to calcification of the cardiovascular system and affecting the vital prognosis.
Product Summary of UPASITA® IV Injection Syringe for Dialysis
Brand name:
UPASITA® IV Injection Syringe for Dialysis 25μg
UPASITA® IV Injection Syringe for Dialysis 50μg
UPASITA® IV Injection Syringe for Dialysis 100μg
UPASITA® IV Injection Syringe for Dialysis 150μg
UPASITA® IV Injection Syringe for Dialysis 200μg
UPASITA® IV Injection Syringe for Dialysis 250μg
UPASITA® IV Injection Syringe for Dialysis 300μg
Generic Name (JAN):
Upacicalcet Sodium Hydrate
Date of Marketing Approval:
June 23, 2021
Indications:
Secondary hyperparathyroidism in patients on hemodialysis
Dosage and Administration:
In adults, UPASITA® is usually administered into venous line of the dialysis circuit at the end of dialysis session during rinse back at a dose of 25 μg sodium upacicalcet 3 times a week as a starting dose.
The starting dose can be 50 μg depending on the concentration of serum calcium. Thereafter, the dose may be adjusted in a range from 25 to 300 μg while parathyroid hormone (PTH) and serum calcium level should be carefully monitored in patients.
SYN
WO 2020204117
PATENT
WO 2011108724
WO 2011108690
JP 2013063971
WO 2016194881
JP 6510136
PATENT
WO 2016194881
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016194881&tab=FULLTEXT
(2S) -2-amino-3-{[(5-chloro-2-hydroxy-3-sulfophenyl) carbamoyl] amino} propanoic acid (Compound 1 )
[Chemical formula 14]
1H-NMR (400MHz, DMSO-d6): δ 8.3 (s, 1H), 8.2 (bs, 3H), 8.1 (d, 1H, J = 2.6Hz), 7.3 (t, 1H, J = 6.0Hz), 7.0 (d, 1H, J = 2.6Hz), 4.0-4.1 (m, 1H), 3.6-3.7 (m, 1H), 3.4-3.5 (m, 1H)
(2S) -2-amino-3-{[(3-sulfophenyl) carbamoyl] amino} propanoic acid (Compound 2 )
[Chemical
1H-NMR (400MHz, DMSO-d6): δ 8.8 (s, 1H), 8.2 (bs, 3H), 7.7 (s, 1H), 7.3-7.4 (m, 1H), 7.1-7.2 (m, 2H) , 6.3-6.4 (bs, 1H), 4.0-4.1 (bs, 1H), 3.6-3.7 (bs, 1H), 3.5-3.6 (bs, 1H)
(2S) -2-amino-3-{[(3-chloro-2-methyl-5-sulfophenyl) carbamoyl] amino} propanoic acid (Compound 3 )
[Chemical formula 16]
1H-NMR (400MHz, DMSO-d6): δ 14.70-13.30 (bs, 1H), 8.27 (bs, 3H), 8.15 (s, 1H), 7.98 (d, 1H, J = 1.6Hz), 7.27 (d , 1H, J = 1.6Hz), 6.82 (t, 1H, J = 6.0Hz), 4.04 (bs, 1H), 3.70-3.60 (m, 1H), 3.60-3.50 (m, 1H), 2.22 (s, 3H)
compound 3 using phenylchloroformate as a carbonyl group-introducing reagent
(Step 1)
[Chemical
30 mL of water was added and concentrated to 77.0 g at 40 ° C. and 5 kPa. After 100 mL (10 L / kg) of AcOEt was added and the liquid separation operation was performed, 30 mL of water was added to the organic layer and the liquid separation operation was performed again. The organic layer was concentrated to 47.6 g at 40 ° C. and 10 kPa, and then 15 mL (1.5 L / kg) of AcOEt and 100 mL (10 L / kg) of THF were added. Again, it was concentrated to 50.7 g and THF was added up to 146 g. When it was concentrated again to 35.5 g and added to AcOEt 30 mL (3 L / kg) and THF 100 mL (10 L / kg), a solid was precipitated. It was cooled to 5 ° C. and aged overnight. The precipitated solid was filtered under reduced pressure, washed with 20 mL (2.0 L / kg) of THF, and then dried under reduced pressure at 40 ° C. for 3 hours overnight at 30 ° C. to obtain 24.9 g of the desired product as a white solid (net). 23.0 g, 83.6%).
1 H-NMR (400MHz, DMSO-d6): δ 8.86 (bs, 1H), 8.09 (s, 1H), 7.88 (s, 1H), 7.25 (d, 1H, J = 1.6Hz), 7.14 (d, 1H, J = 7.6Hz), 6.60 (t, 1H, J = 5.6Hz), 4.00-3.90 (m, 1H), 3.60-3.50 (m, 1H), 3.30-3.20 (m, 1H), 3.15-3.05 (m, 6H), 2.19 (s, 3H), 1.50-1.30 (m, 18H), 1.20-1.10 (m, 9H)(Step 2)
[Chemical
formula 18] Compound 4 21.64 g (net. 20.0 g, 68 mL of water (3.4 L / kg vs. compound 4) vs. 32.8 mmol) ) Was added, the mixture was stirred at 50 ° C., and 12 mL (135.6 mmol, 4.1 eq.) Of concentrated hydrochloric acid was added dropwise. After stirring for 1 hour, the temperature was raised to 70 ° C. to dissolve the precipitated solid. After confirming the completion of the reaction by HPLC, the mixture was cooled to 50 ° C. and aged for 1 hour, and then cooled to 5 ° C. over 4 hours. The precipitated solid was filtered under reduced pressure, washed with 40 mL (2.0 L / kg) of MeCN / water (2/1), and then dried under reduced pressure at 60 ° C. for 3 hours to obtain 11.2 g of the desired product as a white solid (11.2 g). net 10.5 g, 91.1%).
[Chemical
(Step 1)
3-({[(2S) -2-amino-3-methoxy-3-oxopropyl] carbamoyl} amino) -5-chloro-4-methylbenzene-1-sulfonic acid ( Synthesis of Compound 5 )
[Chemical formula 20] To
1H-NMR (400MHz, DMSO-d6): δ 8.39 (bs, 3H), 8.16 (d, 1H, J = 1.2Hz), 7.90 (d, 1H, J = 1.6Hz), 7.28 (d, 1H, J = 1.6Hz), 6.78 (t, 1H, J = 5.6Hz), 4.20-4.10 (m, 1H), 3.77 (s, 3H), 3.70-3.60 (m, 1H), 3.55-3.45 (m, 1H) , 2.21 (s, 3H)
HRMS (FAB – ): calcd for m / z 364.0369 (MH), found The m / z 364.0395 (MH)(step 2)
[Formula 21]
compound 5 10.64 g (net Non 10.0 g, To 27.34 mmol), 18 mL of water (1.8 L / kg vs. compound 5 ) was added and stirred at 8 ° C. 3.42 mL (57.41 mmol, 2.1 eq.) Of a 48% aqueous sodium hydroxide solution was added dropwise, and the mixture was washed with 1.0 mL (1.0 L / kg) of water and then stirred at 8 ° C. for 15 minutes. After confirming the completion of hydrolysis by HPLC, the temperature was raised to 25 ° C. and 48% HBr aq. The pH was adjusted to 5.8 by adding about 3.55 mL. After confirming the precipitation of the target product by dropping 65 mL (6.5 L / kg) of IPA, the mixture was aged for 1 hour. 81 mL (8.1 L / kg) of IPA was added dropwise and aged at 8 ° C. overnight. The precipitated solid was filtered under reduced pressure, washed with 20 mL (2.0 L / kg) of IPA, and then dried under reduced pressure at 40 ° C. for 4 hours to obtain 10.7 g of the desired product as a white solid (net 9.46 g, 92. 6%).
1 H-NMR (400MHz, DMSO-d6): δ8.76 (s, 1H), 7.91 (d, 1H, J = 1.6Hz), 8.00-7.50 (bs, 2H), 7.24 (d, 1H, J = 1.6Hz), 7.20 (t, 1H, J = 5.6Hz), 3.58-3.54 (m, 1H), 3.47-3.43 (m, 1H), 3.42-3.37 (m, 1H), 2.23 (s, 3H)
(Step 1)
[Chemical
1 H-NMR (400MHz, DMSO-d6): δ 9.76 (bs, 1H), 8.93-8.90 (m, 2H), 8.60-8.50 (m, 1H), 8.10-8.00 (m, 2H), 7.60 (s , 1H), 7.50-7.40 (m, 3H), 7.30-7.20 (m, 3H), 2.30 (s, 3H)(Step 2)
[Chemical 23]
Compound 6 To 5.0 g (11.9 mmol), 50 ml of acetonitrile and 3.53 g (11.9 mmol) of Boc-DAP-OtBu were added, and the mixture was stirred at 8 ° C. 3.5 ml (25 mmol) of triethylamine was added dropwise, and the mixture was stirred overnight at room temperature. The solvent was distilled off under reduced pressure, and 25 ml of ethyl acetate and 5 ml of water were added for extraction. The organic layer was washed with 5 ml of water, the solvent was distilled off, 50 ml of tetrahydrofuran was added, the mixture was cooled to 8 ° C., and aged for 1 hour. The precipitated solid was filtered under reduced pressure, washed with 10 ml of tetrahydrofuran, and dried under reduced pressure at 60 ° C. overnight to obtain 6.3 g of the desired product as a white solid.
[Chemical
using phenyl chloroformate as a carbonyl group introduction reagent [Chemical formula 25] MeCN 73 mL (14.6 L) with respect to 5.00 g (22.4 mmol) of ACHB. / Kg vs ACHB), Py 3.8 mL (47 mmol, 2.1 eq.), Was added and stirred at 40 ° C. After adding 3.0 mL (24 mmol, 1.05 eq.) Of ClCO 2 Ph and stirring for 30 minutes, the completion of the CM conversion reaction was confirmed by HPLC. 5.87 g (23 mmol, 1.0 eq.) Of Boc-DAP-OMe was added, washed with a small amount of MeCN, 9.7 mL (70 mmol, 3.1 eq.) Of TEA was added dropwise, and the mixture was stirred at 40 ° C. for 3 hours. After confirming the completion of the urea conversion reaction by HPLC, the mixture was cooled to room temperature. 7.3 mL (112 mmol, 5.0 eq.) Of MsOH was added, the temperature was raised to 50 ° C., and the mixture was stirred for 7 hours. Further, 1.5 mL (23 mmol, 1.0 eq.) Of MsOH was added, and the reaction was carried out at 50 ° C. overnight. After confirming the completion of deprotection by HPLC, 90 mL of acetone was added to the reaction solution, and the mixture was cooled to room temperature. The precipitated solid was obtained and dried under reduced pressure at 60 ° C. to obtain the desired product. 1 H-NMR (400MHz, DMSO-d6): δ 7.22 (m, 1H), 7.14 (m, 1H), 4.36 (m, 1H), 3.80 (s, 3H), 3.20-3.40 (m, 2H).
compound 5 using 4-chlorophenylchloroformate as a carbonyl group-introducing reagent
[Chemical formula 26] For
compound 5 using 4-nitrophenyl chloroformate as a carbonyl group-introducing reagent
[Chemical formula 27]
compound 3 using Boc-DAP-OH
[Chemical 28]
PATENT
JP 6510136
PATENT
WO 2020204117
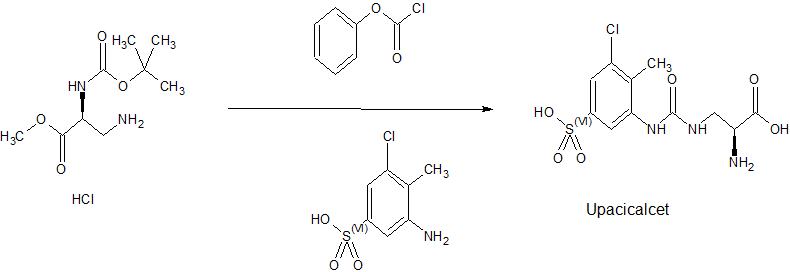
Synthesis of 3-{[(2S) -2-amino-2-carboxyethyl] carbamoylamino} -5-chloro-4-methylbenzenesulfonate sodium (Compound A1)
Synthesis of
3 -({[(2S) -2-amino-3-methoxy-3-oxopropyl] carbamoyl} amino) -5-chloro-4-methylbenzene-1-sulfonic acid 3-amino- 37.5 mL (7.5 L / kg vs ACTS) of acetonitrile and 3.81 mL (47.38 mmol, 2.1 eq.) Of pyridine against 5 g (22.56 mmol) of 5-chloro-4-methylbenzenesulfonic acid (ACTS). Was added and stirred at 25 ° C. 2.99 mL (23.68 mmol, 1.05 eq.) Of ClCO 2 Ph was added dropwise, and after stirring for 30 minutes, the completion of the carbamate reaction was confirmed by HPLC. Add 5.92 g (23.23 mmol, 1.03 eq.) Of 3-amino-N- (tert-butoxycarbonyl) -L-alanine methyl ester hydrochloride and 9.75 mL (69.93 mmol, 3.1 eq.) Triethylamine. Was added dropwise, and the mixture was stirred at 25 ° C. for 3 hours. Add 0.4 g (1.58 mmol, 0.07 eq.) Of 3-amino-N- (tert-butoxycarbonyl) -L-alanine methyl ester hydrochloride and 0.22 mL (1.58 mmol, 0.07 eq.) Of triethylamine. Then, the completion of the urea conversion reaction was confirmed by HPLC. 7.32 mL (112.8 mmol, 5.0 eq.) Of methanesulfonic acid was added, the temperature was raised to 50 ° C., and the mixture was stirred for 4 hours. After confirming the completion of deprotection by HPLC, the mixture was cooled to 25 ° C. and 37.5 mL (7.5 L / kg) of acetonitrile and 7.5 mL (1.5 L / kg) of water were added to precipitate a solid. It was cooled to 5 ° C. and aged for 16 hours. The precipitated solid was filtered under reduced pressure, washed with 20 mL (4.0 L / kg) of water / acetonitrile (1/2), and then dried under reduced pressure at 40 ° C. for 5 hours to obtain 7.72 g of the desired product as a white solid (. net 7.20 g, 87.3%).
1 H-NMR (400MHz, DMSO-d6): δ 8.39 (bs, 3H), 8.16 (d, 1H, J = 1.2Hz), 7.90 (d, 1H, J = 1.6Hz), 7.28 (d, 1H, J = 1.6Hz), 6.78 (t, 1H, J = 5.6Hz), 4.20-4.10 (m, 1H), 3.77 (s, 3H), 3.70-3.60 (m, 1H), 3.55-3.45 (m, 1H) ), 2.21 (S, 3H)
(2)
Compound obtained in step 1 of synthesis of 3-{[(2S) -2-amino-2-carboxyethyl] carbamoylamino} -5-chloro-4-methylbenzenesulfonate . To 64 g (net 10.0 g, 27.34 mmol), 18 mL of water (1.8 L / kg vs. the compound of Step 1) was added, and the mixture was stirred at 8 ° C. 3.42 mL (57.41 mmol, 2.1 eq.) Of a 48% aqueous sodium hydroxide solution was added dropwise, and the mixture was washed with 1.0 mL (1.0 L / kg) of water and then stirred at 8 ° C. for 15 minutes. After confirming the completion of hydrolysis by HPLC, the temperature was raised to 25 ° C. and 48% HBr aq. About 3.55 mL was added to adjust the pH to 5.8. After confirming the precipitation of the desired product by dropping 65 mL (6.5 L / kg) of isopropyl alcohol, the mixture was aged for 1 hour. 81 mL (8.1 L / kg) of isopropyl alcohol was added dropwise and the mixture was aged at 8 ° C. overnight. The precipitated solid was filtered under reduced pressure, washed with 20 mL (2.0 L / kg) of isopropyl alcohol, and then dried under reduced pressure at 40 ° C. for 4 hours to obtain 10.7 g of the desired product as a white solid (net 9.46 g, 92). .6%).
///////////Upacicalcet sodium hydrate, Upasita, ウパシカルセトナトリウム水和物 , APPROVALS 2021, JAPAN 2021, Upacicalcet