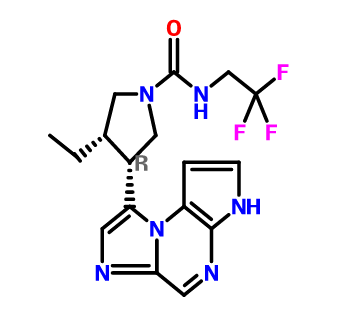
ABT 494
(-)-(3S,4R) cis form
CAS 1310726-60-3 FREE FORM
| MF | C17H19F3N6O |
|---|---|
| MW | 380.36757 g/mol |
Tartrate form
C17 H19 F3 N6 O . C4 H6 O6 . 4 H2 O ………….CAS 1607431-21-9
1-Pyrrolidinecarboxamide, 3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)-, (3S,4R)-, (2R,3R)-2,3-dihydroxybutanedioate, hydrate (1:1:4)
FREE FORM
(3s,,4R)-3-ethyl-4-(3H-imidazo[l,2-fl]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine- l-carboxamide.
(35,,4R)-3-ethyl-4-(3H- imidazo[l,2-fl]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine- l-carboxamide,
(cis,)-3-ethyl-4-(3H-imidazo[l,2-fl]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-l-carboxamide
1-Pyrrolidinecarboxamide, 3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)-, (3S,4R)-
rel-(-)-(3S,4R)-3-Ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide
A Jak1 inhibitor potentially for the treatment of rheumatoid arthritis.
pharmaceutically acceptable salts thereof, stereoisomers thereof, and isomers thereof, is provided in U.S. Patent No. 8,426,411,
Abbott Laboratories ABBOTT ……INNOVATOR
Upadacitinib (code name ABT-494) is a drug which is currently under investigation for the treatment of rheumatoid arthritis, Crohn’s disease, ulcerative colitis, and psoriatic arthritis. It was developed by the biotech company AbbVie.
Upadacitinib tartrate, a selective Jak1 inhibitor, is in phase III clinical trials at AbbVie (previously Abbott) for the treatment of patients with moderate to severe rheumatoid arthritis or active psoriatic arthritis with inadequate responses to conventional or biologic disease-modifying antirheumatic drugs (DMARDs). Phase III clinical trials are also ongoing for the treatment of moderately to severely active Crohn’s disease, ulcerative colitis, moderate to severe atopic dermatitis, and active ankylosing spondylitis.
In 2015, orphan drug designation was assigned to the compound for the treatment of pediatric juvenile idiopathic arthritis (JIA) categories excluding systemic JIA. In 2017, additional orphan drug designation was assigned in the U.S. for the treatment of pediatric systemic JIA.
In January 2013, Abbott spun-off its research-based pharmaceutical business into a newly-formed company AbbVie.
Upadacitinib tartrate [USAN]
1607431-21-9
Mechanism of action
The Janus kinases (JAKs) are a family of cytoplasmic tyrosine kinases whose function is to transduce cytokine-mediated signals via the JAK-STAT pathway. There are four JAK subtypes, each of which has overlapping receptor responsibilities. Inhibitors of this enzyme family (jakinibs) have shown efficacy in treating certain inflammatory and autoimmune diseases such as rheumatoid arthritis and Crohn’s disease. However, the first generation of these drugs, tofacitinib and ruxolitinib, lacked subtype selectivity, affecting JAK1/JAK3 and JAK1/JAK2 respectively. This has led to dose-limiting side effects in this otherwise promising class of drugs.[2][3] Upadacitinib is a second generation Janus kinase inhibitor that is selective for the JAK1 subtype of this enzyme over the JAK2 (74-fold), JAK3 (58-fold) and TYK2 subtypes.[4]
Clinical trials
Phase I studies
A phase I study revealed that upadacitinib followed a bi-exponential disposition with a terminal half-life of 6–16 hours.[1] There was no significant accumulation over the dose range of 3–36 mg per day. No interaction was found in rheumatoid arthritis patients taking methotrexate. The most common adverse event was headache but its incidence was similar to that when taking placebo (15.6% for upadacitinib vs. 16.7% for placebo). An investigation into absorption and metabolism found that dosing after a high-fat meal had no effect on upadacitinib total drug exposure over time (area under the curve or AUC).[5] Inhibition of CYP3A by ketoconazole increased total AUC, indicating the importance of this metabolic route.
Phase II studies
Two phase IIb studies were initiated to study the efficacy and safety of upadacitinib in patients with rheumatoid arthritis and one phase II study was initiated in patients with Crohn’s disease.
BALANCE I
In the first study, 276 rheumatoid arthritis patients were recruited who had previously experienced inadequate response to anti–tumor necrosis factor (TNF) therapy and were currently on a stable dose of methotrexate.[6] Patients were randomized to receive 3, 6, 12, or 18 mg twice daily or placebo. The primary endpoint was a 20% improvement in symptoms according to the American College of Rheumatology improvement criteria (ACR20). At the completion of the study it was found that response rates were significantly higher in those receiving upadacitinib versus in those receiving placebo alone (36–42% and 22– 26%, respectively). Adverse events included headache, nausea, and infection but no infections were serious.
BALANCE II
In the second phase IIb study, 300 rheumatoid arthritis patients were recruited who have had an inadequate response to methotrexate.[7] Patients were randomized to receive 3, 6, 12, or 18 mg twice daily or placebo. The primary endpoint was a 20% improvement in symptoms according to the American College of Rheumatology improvement criteria (ACR20). At the completion of the study it was found that response rates were significantly higher in those receiving upadacitinib versus in those receiving placebo alone. (62%, 68%, 80%, 64%, and 76% for the 3, 6, 12, 18, and 24 mg doses, respectively) than with placebo (46%). Improvement in symptoms was rapid, with significant changes in disease scores by week 2. Adverse events were mild with infection being the most serious. One case of community-acquired pneumonia occurred at 12 mg.
CELEST
In this 16-week study, 220 patients were recruited with moderately to severely active Crohn’s disease. Participants must have also experienced an inadequate response to or intolerance to Immunotherapy or TNF inhibitors.[8][9] Patients were randomized to therapy with upadacitinib at 3, 6, 12, 24 mg twice daily or 24 mg once daily for 16 weeks or placebo, followed by blinded extension therapy for 36 weeks. The co-primary endpoints were the proportion of patients who achieved clinical remission (soft stool frequency or daily abdominal pain score) at week 16 and endoscopic remission at week 12 or 16. Secondary endpoints included significant clinical response (≥30% reduction in symptoms) at week 16 and endoscopic response (≥25% decrease in symptoms) at week 12 or 16. At 16 weeks 22% of patients taking the 24 mg twice daily dose achieved endoscopic remission with upadacitinib compared to 0% of patients taking placebo. 27% of patients taking the 6 mg twice daily dose achieved clinical remission compared to 11% of patients taking placebo. Adverse events did not appear to be dose-related. A single case of non-melanoma skin cancer was reported in the 24 mg twice daily group.
Phase III studies
Abbvie has planned a total of six phase III trials that will evaluate over 4,000 patients with moderate to severe rheumatoid arthritis.[10] Two phase III trials are planned studying patients with psoriatic arthritis and one in patients with ulcerative colitis.

PATENT
The synthesis of the compounds of the invention, including (35,,4R)-3-ethyl-4-(3H-imidazo[l,2-fl]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine- l-carboxamide, pharmaceutically acceptable salts thereof, stereoisomers thereof, and isomers thereof, is provided in U.S. Patent No. 8,426,411, the entire content of which is incorporated herein by reference.
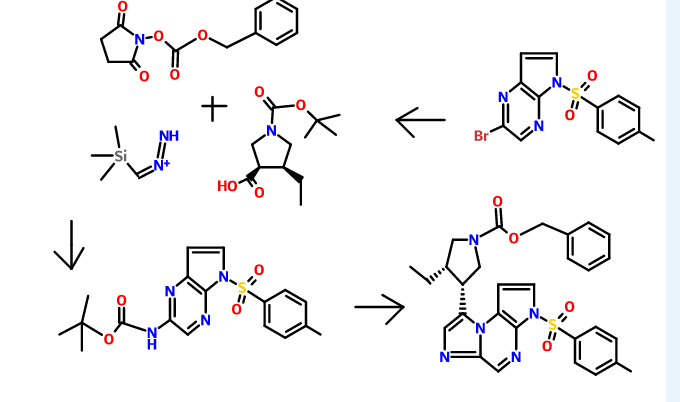
For example, (3lS,,4R)-3-ethyl-4-(3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-l-carboxamide can be synthesized according to the following scheme:
N-Alkylation using alkyl halide, a-haloketone or oc-haloamide
A round bottom flask is charged with a base such as NaH (60% dispersion in mineral oil), K2CO3, or CS2CO3 (preferably NaH (60% dispersion in mineral oil), 0.9-1.5 equiv., preferably 0.95 equiv.) and an organic solvent (such as N, N-dimethylformamide (DMF), dichloromethane (DCM), 1,4-dioxane, or N-methyl-2-pyrrolidone (NMP), preferably DMF). The mixture is cooled to about -10 °C to ambient temperature (preferably about 0°C) and a solution of an appropriately substituted amine (preferably 1 equiv.) in an organic solvent (such as DMF) is added. Alternatively, the base may be added portionwise to a solution of the amine and an organic solvent at about 0°C to ambient temperature. The reaction mixture is stirred for about 5-90 min (preferably about 15-30 min) at about -10°C to ambient temperature (preferably about 0°C) followed by the addition of an alkyl halide, a-haloketone, or cc-haloamide (1-2 equiv., preferably 1.2 equiv.). Alternatively, a solution of an amine and a base in an organic solvent may be added to a solution of an alkyl halide, α-haloketone, or a-haloamide in an organic solvent at about 0°C. The reaction mixture is stirred at about -10°C to ambient temperature (preferably ambient temperature) for about 0.5-24 h (preferably about 1 h). Optionally, the organic solvent may be removed under reduced pressure.
Optionally, the reaction mixture or residue may be diluted with water, aqueous NH4CI, or aqueous NaHC03. If a precipitate forms the solid may be optionally collected via vacuum filtration to give the target compound. Alternatively, an organic solvent (such as ethyl acetate (EtOAc) or DCM) is added to the aqueous mixture and the layers are separated. The aqueous layer may optionally be extracted further with an organic solvent (such as EtOAc and/or DCM). The combined organic layers are optionally washed with additional aqueous solutions such as brine, dried over anhydrous Na2S04 or MgS04, filtered, and concentrated to dryness under reduced pressure.
The procedure above is illustrated below in the preparation of ie/t-butyl 2-amino-2-oxoethyl(5-tosyl-5H-pyrrolo[3,2-b]pyrazin-2-yl)carbamate from ie/t-butyl (5-tosyl-5H-pyrrolo[2,3-b]pyrazin-2-yl)carbamate.
To a solution of iert-butyl 5-tosyl-5H-pyrrolo[3,2-b]pyrazin-2-ylcarbamate (1.00 g, 2.57 mmol, Example #3 Step E) and DMF (13 mL) under nitrogen at about 0 °C was added NaH (60% dispersion in mineral oil, 0.113 g, 2.83 mmol) in one portion. After about 30 min, 2-bromoacetamide (0.391 g, 2.83 mmol) was added in one portion. After about 30 min, the ice bath was removed and the solution was stirred at ambient temperature for about 2 h. Saturated aqueous NH4Cl/water (1: 1, 100 mL) was added. After stirring for about 10 min, the mixture was filtered using water to wash the filter cake. The aqueous phase was extracted with EtOAc (50 mL). The filter cake was dissolved in EtOAc and added to the organic layer. The organic layer was dried over Na2S04, filtered, and concentrated under reduced pressure. The material was purified by silica gel chromatography eluting with a gradient of 20-100% EtOAc/heptane to give tert-butyl 2-amino-2-oxoethyl(5-tosyl-5H-pyrrolo[3,2-b]pyrazin-2-yljcarbamate (0.980 g, 82%): LC/MS (Table 1, Method n) Rt = 0.70 min; MS m/z 446 (M+H)+.
Similar reaction condition can also be used to synthesize benzyl 3-ethyl-4-(2-((5-tosyl-5H-pyrrolo[2,3-b]pyrazin-2-yl)amino)acetyl)pyrrolidine-l-carboxylate from iert-butyl (5-tosyl-5H-pyrrolo[2,3-b]pyrazin-2-yl)carbamate and benzyl 3-(2-bromoacetyl)-4-ethylpyrrolidine- 1 -carboxylate.
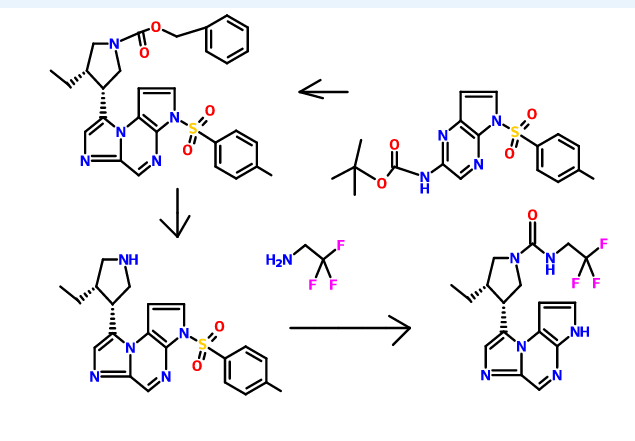
Cyclization of a ketone using a dithiaphosphetane reagent (e.g., synthesizing (3S,4R)-benzyl 3-ethyl-4-(3-tosyl-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazin-8-yl)pyrrolidine-l-carboxylate from benzyl 3-ethyl-4-(2-((5-tosyl-5H-pyrrolo[2,3-Z>]pyrazin-2-yl)amino)acetyl)pyrrolidine-l-carboxylate)
To a solution of a ketone (preferably 1 equiv.) in an organic solvent such as tetrahydrofuran (THF) or 1,4-dioxane (preferably 1,4-dioxane) is added a thiolating reagent such as Lawesson’s reagent or Belleau’s reagent (2,4-bis(4-phenoxyphenyl)-l,3-dithia-2,4-diphosphetane-2,4-disulfide) (0.5-2.0 equiv., preferably Lawesson’s reagent, 0.5-0.6 equiv.). The reaction is heated at about 30°C to 120°C (preferably about 60-70°C) for about 0.5-10 h (preferably about 1-2 h). Optionally, additional thiolating reagent (0.5-2.0 equiv., preferably 0.5-0.6 equiv.) can be added to the reaction mixture and heating can be continued for about 0.5-10 h (preferably about 1-2 h). The reaction mixture is concentrated under reduced pressure.
Preparation of 8-((ds)-4-ethylpyrrolidin-3-yl)-3-tosyl-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazine from (3S,4R)-benzyl 3-ethyl-4-(3-tosyl-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazin-8-yl)pyrrolidine-l-carboxylate
To a solution of (cis)-benzyl 3-ethyl-4-(3-tosyl-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazin-8-yl)pyrrolidine-l-carboxylate (0.838 g, 1.541 mmol) is added a solution of HBr (2.50 mL, 15.19 mmol, 33% in acetic acid). The reaction mixture is stirred at ambient temperature for about 1 h. The reaction is diluted with diethyl ether or Et20 (50 mL) and water (20 mL). The layers are stirred for about 3 min and the organic layer is decanted then the procedure is repeated 5 times. The aqueous layer is cooled to about 0°C and is basified with saturated aqueous NaHC03 solution (10 mL) to about pH 7. The aqueous layer is extracted with EtOAc (3 x 50 mL), combined, and dried over anhydrous Na2S04, filtered and concentrated to give a brown solid. The solid is dissolved in DCM (50 mL) and washed with water (3 x 20 mL), dried over anhydrous Na2S04, filtered and concentrated to afford 8-((cis)-4-ethylpyrrolidin-3-yl)-3-tosyl-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazine (0.453, 61%) as a brown residue: LC/MS (Table 1, Method a) Rt = 1.73 min; MS m/r. 410 (M+H)+.
Hydrolysis of a sulfonamide (e.g., 8-((3R,4S)-4-ethylpyrrolidin-3-yl)-3-tosyl-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazine to 8-((3R,4S)-4-ethylpyrrolidin-3-yl)-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazine)
To a flask containing a sulfonamide, for example, a sulfonyl-protected pyrrole, (preferably 1 equiv.) in an organic solvent (such as 1,4-dioxane, methanol (MeOH), or THF/MeOH, preferably 1,4-dioxane) is added an aqueous base (such as aqueous Na2C03 or aqueous NaOH, 1-30 equiv., preferably 2-3 equiv. for aqueous NaOH, preferably 15-20 equiv. for aqueous Na2C03). The mixture is stirred at about 25-100 °C (preferably about 60 °C) for about 1-72 h (preferably about 1-16 h). In cases where the reaction does not proceed to completion as monitored by TLC, LC/MS, or HPLC, additional aqueous base (such as aqueous Na2C03, 10-20 equiv., preferably 10 equiv. or aqueous NaOH, 1-5 equiv., preferably 1-2 equiv.) and/or a cosolvent (such as ethanol (EtOH)) is added. The reaction is continued at about 25-100°C (preferably about 60°C) for about 0.25-3 h (preferably about 1-2 h). In any case where an additional base labile group is present (for example, an ester a
trifluoromethyl, or a cyano group), this group may also be hydrolyzed. The reaction is worked up using one of the following methods. Method 1. The organic solvent is optionally removed under reduced pressure and the aqueous solution is neutralized with the addition of a suitable aqueous acid (such as aqueous HC1). A suitable organic solvent (such as EtOAc or DCM) and water are added, the layers are separated, and the organic solution is dried over anhydrous Na2S04 or MgS04, filtered, and concentrated to dryness under reduced pressure to give the target compound. Method 2. The organic solvent is optionally removed under reduced pressure, a suitable organic solvent (such as EtOAc or DCM) and water are added, the layers are separated, and the organic solution is dried over anhydrous Na2S04 or MgS04, filtered, and concentrated to dryness under reduced pressure to give the target compound. Method 3. The reaction mixture is concentrated under reduced pressure and directly purified by one of the subsequent methods.
Formation of a urea using CDI or thiocarbonyldiimidazole, respectively (e.g., from 8-((3R,45)-4-ethylpyrrolidin-3-yl)-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazine to (35,4R)-3-ethyl-4-(3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-l-carboxamide)
To a solution or slurry of an amine or amine salt (1-3 equiv., preferably 1-2 equiv.) in an organic solvent such as DCM, THF, or DMF (preferably DMF) at about 20 – 80 °C (preferably about 65 °C) is optionally added an organic base, such as triethylamine (TEA), N,N-diisopropylethylamine (DIEA), pyridine (preferably TEA) (1-10 equiv., preferably 1-5 equiv.) followed by CDI or 1,1 ‘-thiocarbonyldiimidazole (0.5-2 equiv., preferably 1 equiv.). After about 0.5-24 h (preferably about 1-3 h), a second amine or amine salt (1-10 equiv., preferably 1-3 equiv.) is added neat or as a solution or slurry in an organic solvent such as DCM, THF, or DMF (preferably DMF). The reaction is held at about 20 – 80 °C (preferably about 65 °C ) for about 2 – 24 h (preferably about 3 h). If the reaction mixture is heated, it is cooled to ambient temperature. The reaction mixture is partitioned between an organic solvent (such as EtOAc, DCM or 1,4-dioxane) and an aqueous base (such as saturated aqueous NaHC03 or saturated aqueous Na2C03, preferably saturated aqueous NaHC03). Optionally, the reaction mixture is concentrated under reduced pressure and the residue is partitioned as above. In either case, the aqueous layer is then optionally extracted with additional organic solvent such as EtOAc or DCM. The combined organic layers may optionally be washed with brine and concentrated in vacuo or dried over anhydrous Na2S04 or MgS04 and then decanted or filtered prior to concentrating under reduced pressure to give the target compound. Optionally, the reaction mixture is concentrated under reduced pressure and the residue is directly purified.
Chiral preparative HPLC purification
Chiral purification is performed using Varian 218 LC pumps, a Varian CVM 500 with
switching valves and heaters for automatic solvent, column and temperature control and a Varian 701 Fraction collector. Detection methods include a Varian 210 variable wavelength detector, an in-line polarimeter (PDR-chiral advanced laser polarimeter, model ALP2002) used to measure qualitative optical rotation (+/-) and an evaporative light scattering detector (ELSD) (a PS-ELS 2100 (Polymer Laboratories)) using a 100: 1 split flow. ELSD settings are as follows: evaporator: 46 °C, nebulizer: 24 °C and gas flow: 1.1 SLM. The absolute stereochemistry of the purified compounds was assigned arbitrarily and is drawn as such. Compounds of the invention where the absolute stereochemistry has been determined by the use of a commercially available enantiomerically pure starting material, or a stereochemically defined intermediate, or X-ray diffraction are denoted by an asterisk after the example number.
(ci5,)-3-ethyl-4-(3H-imidazo[l,2-fl]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-l-carboxamide isolated using the above method has an Rt min of 1.52, and m/z ESI+ (M+H)+ of 381.
The starting materials and intermediates of the above synthesis scheme may be obtained using the following schemes:
Preparation of starting material of l-(tert-butoxycarbonyl)-4-ethylpyrrolidine-3-carboxylic acid
Step A: ethyl pent-2-ynoate to (Z)-ethyl pent-2-enoate
To a slurry of Lindlar catalyst (0.844 g, 0.396 mmol) in THF (100 mL) and pyridine (10.00 mL) is added ethyl pent-2-ynoate (5.22 mL, 39.6 mmol). The reaction mixture is sparged with hydrogen for about 10 min and an atmosphere of hydrogen is maintained via balloon. After about 15 h the reaction mixture is filtered through a pad of Celite®, diluted with Et20 (30 mL) and washed with saturated aqueous CuS04 (40 mL), followed by water (40 mL). The organic layer is separated, dried over anhydrous MgS04, filtered, and concentrated in vacuo to provide crude (Z)-ethyl pent-2-enoate (5 g, 98%). 1H NMR (DMSO-d6) δ 1.05 (t, 3H), 1.28 (t, 3H), 2.65 (m, 2H), 4.18 (q, 2 H), 5.72 (m, 1H), 6.21 (m, 1H).
Step B: (ds)-ethyl l-benzyl-4-ethylpyrrolidine-3-carboxylate (from (Z)-ethyl pent-2-enoate and N-benzyl-l-methoxy-N-((trimethylsilyl)methyl)methanamine)
To a solution of N-benzyl-l-methoxy-N-((trimethylsilyl)methyl)methanamine (9.98 mL, 39.0 mmol) and (Z)-ethyl pent-2-enoate (5 g, 39.0 mmol) in DCM (50 mL) is added trifluoroacetic acid (TFA) (0.030 mL, 0.390 mmol) at RT. After about 2 days, the reaction mixture is concentrated in vacuo to provide crude (cis)-ethyl 1 -benzyl-4-ethylpyrrolidine-3- carboxylate (9.8 g, 96%) as an oil. LC/MS (Table 1, Method a) Rt = 1.62 min; MS m/z: 262 (M+H)+.
Step C: ethyl l-benzyl-4-ethylpyrrolidine-3-carboxylate to (ds)-ethyl 4-ethylpyrrolidine-3-carboxylate
A Parr shaker is charged with PdOH2 on carbon (2.243 g, 3.19 mmol) and (cis)-et yl l-benzyl-4-ethylpyrrolidine-3-carboxylate (16.7 g, 63.9 mmol) followed by EtOH (100 mL). The reaction mixture is degassed and purged with hydrogen gas and shaken on the parr shaker at 60 psi for about 4 days at ambient temperature. The reaction mixture is degassed and purged with nitrogen. The suspension is filtered through a pad of Celite® washing with EtOH (~ 900 mL). The solvent is removed under reduced pressure to afford (cis)-ethyl 4-ethylpyrrolidine-3 -carboxylate (8.69 g, 79%) as an oil: LC/MS (Table 1, Method a) Rt = 1.11 min; MS m/z: 172 (M+H)+.
Step D: (ds)-ethyl 4-ethylpyrrolidine-3-carboxylate to (ds)-l-(tert-butoxycarbonyl)-4-ethylpyrrolidine-3-carboxylic acid
To a flask charged with (cis)-et yl 4-ethylpyrrolidine-3-carboxylate (8.69g, 50.7 mmol) is added aqueous HCl (6N, 130 mL, 782 mmol). The solution is heated at about 75°C for about 12 h. aqueous HCl (6N, 100 mL, 599 mmol) is added and stirred at about 80 °C for about 20 h. Aqueous HCl (6N, 100 mL, 599 mmol) is added and continued stirring at about 80 °C for about 20 h. The reaction mixture is cooled to ambient temperature and the solvent is removed under reduced pressure. 1,4-Dioxane (275 mL) and water (50 mL) are added followed by portionwise addition of Na2C03 (13.5 g, 127 mmol). Di-ie/t-butyl dicarbonate (13.3 g, 60.9 mmol) is added and the reaction mixture is stirred at ambient temperature for about 16 h. The solid is filtered and washed with EtOAc (250 mL). The aqueous layer is acidified with aqueous HCl (IN) to about pH 3-4. The layers are partitioned and the aqueous layer is extracted with EtOAc (3 x 100 mL). The combined organic layers are dried over anhydrous Na2S04, filtered and removed under reduced pressure. As the organic layer is almost fully concentrated (~ 10 mL remaining), a solid precipitated. Heptane (30 mL) is added and the solid is filtered washing with heptane to afford (cis)-l-(tert-butoxycarbonyl)-4-ethylpyrrolidine-3-carboxylic acid (3.9 g, 32%) as an off white solid as product: LC/MS (Table 1, Method c) Rt = 0.57 min; MS m/z: 242 (M-H)~.
Synthesis of Intermediate benzyl 3-(2-bromoacetyl)-4-ethylpyrrolidine-l-carboxylate
Acidic cleavage of a Boc-protected amine (e.g., l-(tert-butoxycarbonyl)-4-ethylpyrrolidine-3-carboxylic acid to 4-ethylpyrrolidine-3-carboxylic acid
hydrochloride)
To a solution of a Boc-protected amine (preferably 1 equiv.) in an organic solvent (such as DCM, 1,4-dioxane, or MeOH) is added TFA or HC1 (preferably 4 N HC1 in 1,4-dioxane, 2-35 equiv., preferably 2-15 equiv.). The reaction is stirred at about 20-100 °C (preferably ambient temperature to about 60 °C) for about 1-24 h (preferably about 1-6 h). In any case where an additional acid labile group is present (for example, a t-butyl ester), this group may also be cleaved during the reaction. Optionally, additional TFA or HC1
(preferably 4 N HC1 in 1,4-dioxane solution, 2-35 equiv., preferably 2-15 equiv.) may be added to the reaction mixture in cases where the reaction does not proceed to completion as monitored by TLC, LC/MS, or HPLC. Once the reaction has proceeded to an acceptable level, the reaction mixture can be concentrated in vacuo to provide the amine as a salt.
Alternatively, the reaction may be partitioned between an organic solvent (such as EtOAc, DCM or 1,4-dioxane) and an aqueous base (such as saturated aqueous NaHC03 or saturated aqueous Na2C03, preferably saturated aqueous NaHC03). The aqueous layer can be optionally extracted with additional organic solvent such as EtOAc or DCM. The combined organic layers may optionally be washed with brine, dried over anhydrous Na2S04 or MgS04, then decanted or filtered, prior to concentrating under reduced pressure to give the target compound.
Cbz-protection of an amine (e.g., 4-ethylpyrrolidine-3-carboxylic acid hydrochloride to l-((benzyloxy)carbonyl)-4-ethylpyrrolidine-3-carboxylic acid)
A solution of an amine or an amine salt (preferably 1 equiv.) and a base (for example, Na2C03 or NaOH, 1-3 equiv., preferably Na2C03, 1.6 equiv.) in water or aqueous organic solvent (for example, water / 1,4-dioxane or water / acetonitrile (MeCN), preferably water/ 1,4-dioxane) is stirred at ambient temperature for about 1-10 min (preferably 5 min). A solution of benzyl 2,5-dioxopyrrolidin-l-yl carbonate (1-2 equiv., preferably 1.0 equiv.) in an organic solvent such as 1,4-dioxane or MeCN is added to the reaction. The reaction is stirred at ambient temperature for about 8-144 h (preferably about 72 h). Optionally, the reaction mixture is concentrated under reduced pressure. The resulting aqueous solution is diluted with an organic solvent (such as EtOAc or DCM). The organic extracts are optionally washed with water and/or brine, dried over anhydrous Na2S04 or MgS04, filtered or decanted, and concentrated under reduced pressure. Alternatively, the resulting aqueous solution is acidified by adding an acid such as aqueous NH4C1 or HC1 and is then extracted with an organic solvent (such as EtOAc or DCM).
Formation of a bromomethyl ketone from an acid (e.g., l-((benzyloxy)carbonyl)-4-ethylpyrrolidine-3-carboxylic acid to benzyl 3-(2-bromoacetyl)-4-ethylpyrrolidine-l-carboxylate)
To a solution of a carboxylic acid (preferably 1 equiv.) in an organic solvent (DCM or 1,2-dichloroethane (DCE), preferably DCM) is slowly added oxalyl chloride (1.2-3.0 equiv., preferably 2.2 equiv.) followed by dropwise addition of DMF (0.01-0.20 equiv., preferably about 0.15 equiv.). The reaction is stirred at about 0-40 °C (preferably ambient temperature) for about 3-24 h (preferably about 14 h) before it is concentrated under reduced pressure to a constant weight to give the crude acid chloride. A solution of a crude acid chloride
(preferably 1 equiv.) in an organic solvent (such as THF, MeCN, Et20, or THF/MeCN, preferably THF/MeCN) is added to trimethylsilyldiazomethane (2.0 M in Et20) or diazomethane solution in Et20 (prepared from DIAZALD® according to Aldrich protocol or J. Chromatogr. Sci. 1991, 29:8) (2-10 equiv., preferably 3.5 equiv. of
trimethylsilyldiazomethane) at about -20-20 °C (preferably about 0 °C) in a suitable organic solvent such as THF, MeCN, Et20, or THF/MeCN (preferably THF/MeCN). The reaction mixture is stirred for about 0.5-5 h (preferably about 3 h) at about -20-20 °C (preferably about 0 °C) before the dropwise addition of 48% aqueous HBr (5-40 equiv., preferably about 10 equiv.). After about 0-30 min, (preferably about 5 min) the reaction mixture can be concentrated to dryness to give the desired product, neutralized by a dropwise addition of saturated aqueous NaHC03 or is optionally washed with brine after optional addition of an organic solvent (such as EtOAc or DCM, preferably EtOAc). In cases where the reaction mixture is subjected to an aqueous work-up, the organic layer is dried over anhydrous Na2S04 or MgS04 (preferably MgS04), filtered, and concentrated under reduced pressure.
Synthesis of Intermediate tert-butyl (5-tosyl-5H-pyrrolo[2,3-Z>]pyrazin-2-yl)carbamate
Step A: 3,5-dibromopyrazin-2-amine to 5-bromo-3-((trimethylsilyl)ethynyl)pyrazin-2-amine
To a solution of 3,5-dibromopyrazin-2-amine (125 g, 494 mmol), TEA (207.0 mL, 1483 mmol), and copper (I) iodide (0.941 g, 4.94 mmol) in THF (1255 mL) is added
PdCl2(PPh3)2 (3.47 g, 4.94 mmol). The reaction mixture is cooled at about -5-0°C and a solution of (trimethylsilyl)acetylene (65.0 mL, 470 mmol) in THF (157 mL) is added dropwise over about 15 min. The reaction mixture is stirred at about -5-0°C for about 1.5 h and then allowed to warm to room temperature (RT) overnight. The reaction mixture is then filtered through a CELITE® pad and washed with THF until no further product eluted. The filtrate is concentrated under reduced pressure to give a brown-orange solid. The solid is triturated and sonicated with warm petroleum ether (b.p. 30-60°C, 400 mL), cooled to RT, collected, washed with petroleum ether (b.p. 30-60°C; 2 x 60 mL), and dried to give 5-bmmo-3-((trimethylsilyl)ethynyl)pyrazin-2-amine (124 g, 93%, 93% purity) as a brown solid: LC/MS (Table 1, Method b) Rt = 2.51 min; MS m/z: 270, 272 (M+H)+.
Step B: 5-bromo-3-((trimethylsilyl)ethynyl)pyrazin-2-amine to 2-bromo-5-tosyl-5H-pyrrolo[2,3-Z>]pyrazine
To a solution of 5-bromo-3-((trimethylsilyl)ethynyl)pyrazin-2-amine (3.00g, 11.1 mmol) in DMF (60 mL) at about 0 °C is added NaH (60% dispersion in mineral oil, 0.577g, 14.4 mmol) in three portions. After about 15 min, p-toluenesulfonyl chloride (2.75g, 14.4 mmol) is added and the reaction is allowed to warm slowly to ambient temperature. After about 16 h, the reaction mixture is poured onto ice-cold water (120 mL) and the precipitate is collected by vacuum filtration. The crude solid is dissolved in DCM (15 mL) and purified by silica gel chromatography eluting with DCM to give 2-bromo-5-tosyl-5H-pyrrolo[2,3-bjpyrazine (2.16 g, 52%): LC/MS (Table 1, Method c) Rt = 1.58 min; MS m/z: 352, 354 (M+H)+.
Step C: 2-bromo-5-tosyl-5H-pyrrolo[2,3-b]pyrazine to methyl 5-tosyl-5H-pyrrolo[2,3-Z>]pyrazine-2-carboxylate
CO is bubbled into an orange solution of 2-bromo-5-tosyl-5H-pyrrolo[2,3-b]pyrazine (50. Og, 142 mmol) in DMF (2.50 L) within a 5 L round bottom flask for about 2 min.
Bis(triphenylphosphine)-palladium(II) dichloride (9.96g, 14.2 mmol), TEA (59 mL, 423 mmol) and MeOH (173.0 mL, 4259 mmol) are added and the flask is fitted with a balloon of CO. The mixture is heated at about 95°C under an atmosphere of CO (1 atmosphere). After stirring overnight, the reaction mixture is cooled to ambient temperature overnight and poured into ice water (3.2 L). The mixture is stirred for about 10 min and the precipitate is collected by filtration, while washing with water, and dried for 1 h. The crude material is dissolved in DCM, separated from residual water, dried over anhydrous MgS04, filtered, added silica gel, and concentrated under reduced pressure to prepare for chromatography. The crude material is purified by silica gel column chromatography eluting with 0-5% MeOH in DCM to yield methyl 5-tosyl-5H-pyrrolo[2,3-b]pyrazine-2-carboxylate with 5 mol% DCM as an excipient (40.7 g, 86%, 93% purity): LC/MS (Table 1, Method a) Rt = 2.35 min;
MS m/z 332 (M+H)+.
Step D: methyl 5-tosyl-5H-pyrrolo[2,3-Z>]pyrazine-2-carboxylate to 5-tosyl-5H-pyrrolo[2,3-/>]pyrazine-2-carboxylic acid
HC1 (6 N aqueous, 714 mL) is added to a yellow solution of methyl 5-tosyl-5H-pyrrolo[2,3-b]pyrazine-2-carboxylate (17.8g, 53.6 mmol) in 1,4-dioxane (715 mL) within a 2 L round bottom flask, and the mixture is heated at about 60°C for about 16 h. The reaction mixture is cooled to ambient temperature. The organic solvent is removed under reduced pressure and the precipitate is collected, washed with water, and dried to yield 5-tosyl-5H-pyrrolo[2,3-b]pyrazine-2-carboxylic acid (14.4 g, 85%) as a yellow solid: LC/MS (Table 1, Method a) Rt = 1.63 min; MS m/z 316 (Μ-Η)“.
Step E: 5-tosyl-5H-pyrrolo[2,3-b]pyrazine-2-carboxylic acid to tert-butyl 5-tosyl-5H-pyrrolo[2,3-Z>]pyrazin-2-ylcarbamate
In a 500 mL round bottom flask, 5-tosyl-5H-pyrrolo[2,3-b]pyrazine-2-carboxylic acid (14.4 g, 45.3 mmol), diphenylphosphoryl azide (9.78 mL, 45.3 mmol) and TEA (13.9 mL, 100 mmol) in ie/t-butanol (i-BuOH) (200 mL) are added to give an orange suspension. The mixture is heated at about 70°C for about 16 h, cooled to ambient temperature and the insoluble material is removed by filtration. The solvent is removed under reduced pressure and the crude material is purified by silica gel column chromatography eluting with 25-60% EtOAc in heptane to yield tert-butyl 5-tosyl-5H-pyrrolo[2,3-b]pyrazin-2-ylcarbamate (9.75 g, 54%) as an off-white solid: LC/MS (Table 1, Method a) Rt = 2.79 min; MS m/z 389 (M+H)+.
PATENT
WO2011068881


PATENT
http://www.google.com/patents/US20110311474
Preparation #F.1.1: 8-((cis)-4-ethylpyrrolidin-3-yl)-3-tosyl-3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazine
-

-
To a solution of (cis)-benzyl 3-ethyl-4-(3-tosyl-3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazin-8-yl)pyrrolidine-1-carboxylate (0.838 g, 1.541 mmol, prepared using E from Example #36 Step D with TFA, N, R, S.1 with Example #3 Step E, and T with Lawesson’s reagent) was added a solution of HBr (2.50 mL, 15.19 mmol, 33% in acetic acid). The reaction mixture was stirred at ambient temperature for about 1 h. The reaction was diluted with Et2O (50 mL) and water (20 mL). The layers were stirred for about 3 min and the organic layer was decanted then the procedure was repeated 5 times. The aqueous layer was cooled to about 0° C. was basified with saturated aqueous NaHCO3solution (10 mL) to about pH 7. The aqueous layer was extracted with EtOAc (3×50 mL), combined, and dried over anhydrous Na2SO4, filtered and concd to give a brown solid. The solid was dissolved in DCM (50 mL) and washed with water (3×20 mL), dried over anhydrous Na2SO4, filtered and coned to afford 8-((cis)-4-ethylpyrrolidin-3-yl)-3-tosyl-3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazine (0.453, 61%) as a brown residue: LC/MS (Table 1, Method a) Rt=1.73 min; MS m/z: 410 (M+H)+.
SEE…………..1-((cis)-4-ethylpyrrolidin-3-yl)-6-tosyl-6H-pyrrolo[2,3-e][1,2,4]triazolo[4,3-a]pyrazine (0.250 g, 0.609 mmol, Example #36, step F)
PATENTS
WO-2017066775
WO-2015061665
WO-2011068881
References
- ^ Jump up to:a b c Mohamed, Mohamed-Eslam F.; Camp, Heidi S.; Jiang, Ping; Padley, Robert J.; Asatryan, Armen; Othman, Ahmed A. (December 2016). “Pharmacokinetics, Safety and Tolerability of ABT-494, a Novel Selective JAK 1 Inhibitor, in Healthy Volunteers and Subjects with Rheumatoid Arthritis”. Clinical Pharmacokinetics. 55 (12): 1547–1558. doi:10.1007/s40262-016-0419-y. ISSN 1179-1926. PMID 27272171.
- Jump up^ Fleischmann, Roy (May 2012). “Novel small-molecular therapeutics for rheumatoid arthritis”. Current Opinion in Rheumatology. 24 (3): 335–341. doi:10.1097/BOR.0b013e32835190ef. ISSN 1531-6963. PMID 22357358.
- Jump up^ Riese, Richard J.; Krishnaswami, Sriram; Kremer, Joel (August 2010). “Inhibition of JAK kinases in patients with rheumatoid arthritis: scientific rationale and clinical outcomes”. Best Practice & Research. Clinical Rheumatology. 24 (4): 513–526. doi:10.1016/j.berh.2010.02.003. ISSN 1532-1770. PMID 20732649.
- Jump up^ “Characterization of ABT-494, a Second Generation Jak1 Selective Inhibitor”. ACR Meeting Abstracts. Retrieved 2017-05-21.
- Jump up^ Mohamed, Mohamed-Eslam F.; Jungerwirth, Steven; Asatryan, Armen; Jiang, Ping; Othman, Ahmed A. (2017-05-14). “Assessment of effect of CYP3A Inhibition, CYP Induction, OATP1B Inhibition, and High-Fat Meal on Pharmacokinetics of the JAK1 inhibitor Upadacitinib”. British Journal of Clinical Pharmacology. doi:10.1111/bcp.13329. ISSN 1365-2125. PMID 28503781.
- Jump up^ Kremer, Joel M.; Emery, Paul; Camp, Heidi S.; Friedman, Alan; Wang, Li; Othman, Ahmed A.; Khan, Nasser; Pangan, Aileen L.; Jungerwirth, Steven; Keystone, Edward C. (December 2016). “A Phase IIb Study of ABT‐494, a Selective JAK‐1 Inhibitor, in Patients With Rheumatoid Arthritis and an Inadequate Response to Anti–Tumor Necrosis Factor Therapy”. Arthritis & Rheumatology (Hoboken, N.j.). 68 (12): 2867–2877. doi:10.1002/art.39801. ISSN 2326-5191. PMC 5132116
 . PMID 27389975.
. PMID 27389975. - Jump up^ Genovese, Mark C.; Smolen, Josef S.; Weinblatt, Michael E.; Burmester, Gerd R.; Meerwein, Sebastian; Camp, Heidi S.; Wang, Li; Othman, Ahmed A.; Khan, Nasser; Pangan, Aileen L.; Jungerwirth, Steven (December 2016). “Efficacy and Safety of ABT‐494, a Selective JAK‐1 Inhibitor, in a Phase IIb Study in Patients With Rheumatoid Arthritis and an Inadequate Response to Methotrexate”. Arthritis & Rheumatology (Hoboken, N.j.). 68 (12): 2857–2866. doi:10.1002/art.39808. ISSN 2326-5191. PMC 5132065 . PMID 27390150.
- Jump up^ “A Multicenter, Randomized, Double-Blind, Placebo-Controlled Study of ABT-494 for the Induction of Symptomatic and Endoscopic Remission in Subjects With Moderately to Severely Active Crohn’s Disease Who Have Inadequately Responded to or Are Intolerant to Immunomodulators or Anti-TNF Therapy – Full Text View – ClinicalTrials.gov”. Retrieved 2017-05-22.
- Jump up^ “AbbVie Announces Positive Phase 2 Study Results for Upadacitinib (ABT-494), an Investigational JAK1-Selective Inhibitor, in Crohn’s Disease | AbbVie Newsroom”. Retrieved 2017-05-22.
- Jump up^ Phase 3 upadacitinib trials
-
Upadacitinib 
Clinical data Synonyms ABT-494 Routes of
administrationOral Pharmacokinetic data Metabolism Hepatic (CYP3A major, CYP2D6 minor) [1] Biological half-life 6-15 hours[1] Identifiers CAS Number PubChem CID IUPHAR/BPS ChemSpider UNII ChEMBL Chemical and physical data Formula C17H19F3N6O Molar mass 380.38 g·mol−1 3D model (JSmol)

/////Upadacitinib, ABT 494, упадацитиниб , أوباداسيتينيب , 乌帕替尼 , ORPHAN DRUG, PHASE 3
c21cnc4c(n1c(cn2)[C@@H]3[C@@H](CN(C3)C(=O)NCC(F)(F)F)CC)ccn4
OR
CC[C@@H]1CN(C[C@@H]1c4cnc3cnc2nccc2n34)C(=O)NCC(F)(F)F
Analytical Methods
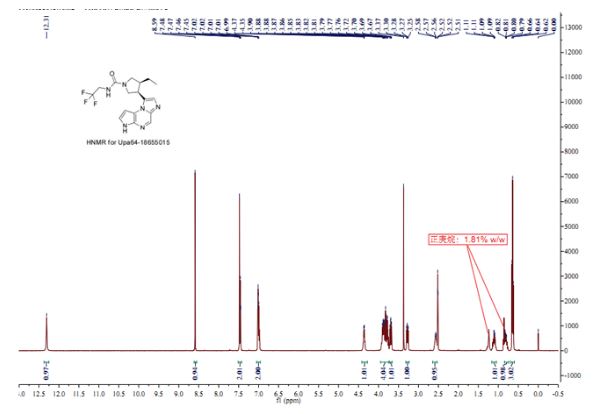
Analytical data was included within the procedures below, in the illustrations of the general procedures, or in the tables of examples. Unless otherwise stated, all !H NMR data were collected on a Varian Mercury Plus 400 MHz or a Varian Inova 600 MHz instrument and chemical shifts
are quoted in parts per million (ppm). LC/MS and HPLC data are referenced to the table of LC/MS and HPLC conditions using the lower case method letter provided in Table 1.
Table 1. LC/MS and HPLC methods
Method Conditions
NH40Ac (pH 4.5) and mobile phase B was HPLC grade MeCN, the column used for the chromatography was a 19 x 50 mm Waters Atlantis T3 OBD CI 8 column (5 μιη particles), detection methods were Photodiode array DAD and Waters ZQ 2000 mass spectrometer.
g HPLC: The gradient was 10% B over 2.5 min then 10-15% B over 0.50 min then 15-75% B over 3 min then 75-85% B over 3.10 min then 85%-100% B over 1.5 min (22.5 mL/min flow rate). Mobile phase A was 50 mM NH4OAc (pH 4.5) and mobile phase B was HPLC grade MeCN, the column used for the chromatography was a 19 x 50 mm Waters Atlantis T3 OBD CI 8 column (5 μιη particles), detection methods were Waters 2996 photodiode array DAD and Waters ZQ 2000 mass spectrometer. h HPLC: The gradient was 10-85% B over 9.00 min then 85-95% B over 0.10 min then held at 95% B for 1.50 min (25.0 mL/min flow rate). Mobile phase A was 50 mM NH4OAc (pH 4.5) and mobile phase B was HPLC grade MeCN, the column used for the chromatography was a 19 x 50 mm Waters Atlantis T3 OBD CI 8 column (5 μιη particles), detection methods were Waters 2996 photodiode array DAD and Waters ZQ 2000 mass spectrometer.
HPLC: The gradient was 10-35% B over 45 min (25 mL/min flow rate). Mobile phase A was 50 mM NH4OAc (pH 4.5) and mobile phase B was HPLC grade MeCN. The column used for the chromatography was a 21.2 x 250 mm Hypersil CI 8 HS column (8 μιη particles). Detection method was UV.
j HPLC: The gradient was 5-75% B over 25 min (21 mL/min flow rate). Mobile phase A was 50 mM NH4OAc (pH 4.5) and mobile phase B was HPLC grade MeCN. The column used for the chromatography was a 21.2 x 250 mm Hypersil CI 8 HS column (8 μιη particles). Detection method was UV.
k HPLC: The gradient was 20-40% B over 30 min (21 mL/min flow rate). Mobile phase A was 50 mM NH4OAc (pH 4.5) and mobile phase B was HPLC grade MeCN. The column used for the chromatography was a 21.2 x 250 mm Hypersil CI 8 HS column (8 μιη particles). Detection method was UV.
1 HPLC: The gradient was 0-100% B over 30 min (21 mL/min flow rate). Mobile phase A was 50 mM NH4OAc (pH 4.5) and mobile phase B was HPLC grade MeCN. The column used for the chromatography was a 21.2 x 250 mm Hypersil CI 8 HS column (8 μιη particles). Detection method was UV.
Method Conditions
m HPLC: The gradient was 5% B, hold for 5 min, 5-100% B over 40 min, hold at 100% for 5 min, 2 min back to 5% B, hold at 5% B for 4 min, (21 mL/min flow rate). Mobile phase A was 50 mM NH4OAc (pH 4.5) and mobile phase B was HPLC grade MeCN. The column used for the chromatography was a 21.2 x 250 mm Hypersil CI 8 HS column (8 μιη particles). Detection method was UV.
n LC/MS: The gradient was 5-60% B in 0.60 min then 60-95% B to 1.0 min with a hold at 95% B for 0.30 min (1.25 mL/min flow rate). Mobile phase A was 10 mM NH4OAc, mobile phase B was HPLC grade MeCN. The column used for the chromatography is 2.1 x 30 mm Acquity UPLC HSS T3 column (1.8 μιη particles). Detection methods are diode array (DAD) and evaporative light scattering (ELSD) detection as well as positive/negative electrospray ionization.
0 LC/MS: The gradient was 60-95% B in 1.15 min with a hold at 95% B for 3 min (1.3 mL/min flow rate). Mobile phase A was 10 mM NH4OAc, mobile phase B was HPLC grade MeCN. The column used for the chromatography is a 4.6 x 50 mm MAC -MOD Halo C8 column (2.7 μιη particles). Detection methods are diode array (DAD) and evaporative light scattering (ELSD) detection as well as
positive/negative electrospray ionization.
P LC/MS: The gradient was 5% B, hold for 0.2 min, 5-95% B over 1.7 min, hold at 95% for 1.3 min, back to 5% B within 0.01 min, (2.3 mL/min flow rate). Mobile phase A was water (0.05% TFA) and mobile phase B was HPLC grade MeCN (0.05% TFA). The column used for the chromatography was a 4.6 x 50 mm XBridge CI 8 column (3.5 μιη particles). Temperature 50 °C. Detection method was UV.
q HPLC: The gradient was 10-75% B over 10 min (22.5 mL/min flow rate). Mobile phase A was 50 mM NH4OAc (pH 4.5) and mobile phase B was HPLC grade MeCN, the column used for the chromatography was a 19 x 50 mm Waters Atlantis T3 OBD CI 8 column (5 μιη particles), detection methods were Photodiode array DAD and Waters ZQ 2000 mass spectrometer.
r LC/MS: The gradient was 80-65% B in 1.80 min then 65-40% B to 2.80 min with a hold at 40% for another 1.20 min (1.3 mL/min flow rate). The column used for the chromatography is a 4.6 x 50 mm X-bridge hilic column (3.5 μιη particles). Mobile phase A was 10 mM ammonium acetate, mobile phase B was HPLC grade acetonitrile. Detection methods are diode array (DAD) and evaporative light scattering (ELSD) detection as well as positive/negative electrospray ionization.
Method Conditions
s HPLC: The gradient was 0-100% B over 15 min, hold at 100% for 15 min (21 mL/min flow rate). Mobile phase A was 50 mM NH4OAc (pH 4.5) and mobile phase B was HPLC grade MeCN. The column used for the chromatography was a 21.2 x 250 mm Hypersil CI 8 HS column (8 μιη particles). Detection method was UV. t HPLC: The gradient was 24% B over 2 min then 24-55% B over 7.6 min then 55- 98%> B over 1 min (25 mL/min flow rate). Mobile phase A was 50 mM NH4OAc (pH 4.5) and mobile phase B was HPLC grade MeCN, the column used for the chromatography was a 19 x 50 mm Waters Atlantis T3 OBD CI 8 column (5 μιη particles), detection methods were Waters 2996 photodiode array DAD and Waters ZQ 2000 mass spectrometer.
u LC/MS: The gradient was 0-0.1 min 10% A, 0.1-1.1 min 10-100% A, 1.1-1.3 min 100% A, then 1.3-1.4 min 100-10% A. Flow rate was 1 mL/min. Mobile phase A was HPLC grade acetonitrile and mobile phase B was 0.1 % trifluoroacetic acid in water. The column used was a Waters BEH C8, 1.7 μιη (2.1mm χ 30mm) at a temperature of 55°C. Detection methods were diode array (DAD) and evaporative light scattering (ELSD) detection as well as positive APCI ionization.
V The gradient was 0-0.1 min 10% A, 0.1-2.6 min 10-100% A, 2.6-2.9 min 100% A, 2.9-3.0 min 100-10% A then 0.5min post-run delay. Flow rate was 2 mL/min. Mobile phase A was HPLC grade acetonitrile and mobile phase B was 0.1 % trifluoroacetic acid in water. The column used for the chromatography was a Phenomenex Luna Combi-HTS C8(2) 5 μιη ΙΟθΑ (2.1 mm χ 50 mm), at a temperature of 55 °C. Detection methods were diode array (DAD) and evaporative light scattering (ELSD) detection as well as positive APCI ionization.
w HPLC: The gradient was 15% B over 3.5 min then 15-46% B over 6.1 min then 46- 98%> B over 1.2 min (25 mL/min flow rate). Mobile phase A was 50 mM NH4OAc (pH 4.5) and mobile phase B was HPLC grade MeCN, the column used for the chromatography was a 19 x 50 mm Waters Atlantis T3 OBD CI 8 column (5 micron particles), detection methods were Waters 2996 photodiode array DAD and Waters ZQ 2000 mass spectrometer.
X HPLC: The gradient was 0-80% B over 5 min, hold at 80% for 4 min, 0.1 min at 90% B, then 90 to 0% B for 2.9 min, hold at 0 % B for 2 min (1 mL/min flow rate). Mobile phase A was 0.1% H3PO4 in water and mobile phase B was HPLC grade MeCN. The column used for the chromatography was a 4.6 x 150 mm Ascentis Express column (2.8 μιη particles). Detection method was UV.
Method Conditions
y HPLC: The gradient was 0-50% B over 45 min (25 mL/min flow rate). Mobile phase A was 50 mM NH4OAc (pH 4.5) and mobile phase B was HPLC grade MeCN. The column used for the chromatography was a 21.2 x 250 mm Hypersil CI 8 HS column (8 μιη particles). Detection method was UV.
Table 2. Chiral HPLC methods
Method Conditions
6 Isocratic 25% A for 18 min (20 mL/min flow rate). Mobile phase A was a 50:50 mixture of HPLC grade MeOH and EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
7 Isocratic 30% A for 18 min (20 mL/min flow rate). Mobile phase A was a 50:50 mixture of HPLC grade MeOH and EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
8 The gradient was 15-54% A in 16 min then step to 90% A in 0.5 min, with a hold at 90% for 4.3 min (20 mL/min flow rate). Mobile phase A was a 50:50 mixture of HPLC grade MeOH and EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
9 The gradient was 10-70% A in 16 min then re-equilibrated at 10% A for 9 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were UV, λ = 315 nm.
10 The gradient was 10-50% A in 19 min with a hold at 50% for 2 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
11 Isocratic 60% A for 20 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were UV, λ = 300 nm.
Method Conditions
12 Isocratic 30% A for 25 min (20 mL/min flow rate). Mobile phase A was HPLC grade IP A, mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
13 Isocratic 20% A for 20 min (20 mL/min flow rate). Mobile phase A was HPLC grade IP A, mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
14 Isocratic 100% EtOH (200 proof) for 20 min (13 mL/min flow rate). The column used for the chromatography was a Daicel IC, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
15 Isocratic 50% A for 20 min (20 mL/min flow rate). Mobile phase A was HPLC grade IP A, mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
16 Isocratic 30% A for 18 min (20 mL/min flow rate). Mobile phase A was a 50:50 mixture of HPLC grade MeOH and EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
17 The gradient was 10-50% A in 19 min with a hold at 50% for 2 min then re- equilibrated at 10% A for 11 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IC, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
18 The gradient was 10-50% A in 19 min with a hold at 50% for 1.5 min (20 mL/min flow rate). Mobile phase A was HPLC grade IP A, mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection Method Conditions
methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
19 The gradient was 10-50% A in 19 min then re-equilibrated at 10% A for 6 min (20 mL/min flow rate). Mobile phase A was HPLC grade IP A, mobile phase B was HPLC grade heptane with 0.1 % diethylamine added. The column used for the chromatography was a Daicel IC, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
20 Isocratic 40% A for 16 min (20 mL/min flow rate). Mobile phase A was HPLC grade IP A, mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
21 Isocratic 40% A for 15-25 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1 % diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
22 The gradient was 10-40% A in 19 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1 % diethylamine added. The column used for the chromatography was a Daicel IB, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
23 The gradient was 15-70% A in 19 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1 % diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
24 Isocratic 15% A for 14 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IC, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
25 Isocratic 30% A for 10 min (20 mL/min flow rate). Mobile phase A was HPLC grade IP A, mobile phase B was HPLC grade heptane with 0.1% diethylamine
Method Conditions
added. The column used for the chromatography was a Daicel IB, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
26 Isocratic 40% A for 5 min then gradient 40 to 95% A in 2 min, with a hold at 95% for 11 min (20 mL/min flow rate). Mobile phase A was a 50:50 mixture of HPLC grade MeOH and EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1 % diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
27 The gradient was 10-50% A in 19 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1 % diethylamine added. The column used for the chromatography was a Daicel IB, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
28 Isocratic 15% A for 35 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
29 The gradient was 10-50% A in 19 min with a hold at 50% for 3 min then re- equilibrate at 10% A for 13 min (1 mL/min flow rate). Mobile phase A was IPA, mobile phase B was HPLC grade heptane with 0.1 % diethylamine added. The column used for the chromatography was a Daicel IC, 4.6 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
30 Isocratic 20% A for 20 min (1 mL/min flow rate). Mobile phase A was IPA, mobile phase B was HPLC grade heptane with 0.1 % diethylamine added. The column used for the chromatography was a Daicel IC, 4.6 x 250 mm column (5 μιη particles). Detection methods were UV, λ = 230 nm as well as positive electrospray ionization.
31 Isocratic 20% A for 10 min (20 mL/min flow rate). Mobile phase A was ethanol (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
Method Conditions
32 The gradient was 10-70% A in 19 min then re-equilibrate at 10% A for 11 min (20 mL/min flow rate). Mobile phase A was ethanol (200 proof), mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were ELSD and optical rotation.
33 Isocratic 30% A for 20-30 min (20 mL/min flow rate). Mobile phase A was ethanol (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
34 Isocratic 40% A for 10-30 min (20 mL/min flow rate). Mobile phase A was ethanol (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IC, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
35 Isocratic 15% A for 22.5 min then step to 60% A and hold for 5 min (20 mL/min flow rate). Mobile phase A was HPLC grade IP A, mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection method was UV, λ = 325 nm
36 Isocratic 40% A for 20 min (20 mL/min flow rate). Mobile phase A was a 50:50 mixture of HPLC grade MeOH and EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1 % diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
37 The gradient was 10-70% A in 19 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IC, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
38 Isocratic 35% A for 25 min (20 mL/min flow rate). Mobile phase A was HPLC grade IP A, mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IC, 20 x 250 mm Method Conditions
column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
39 Isocratic 70% A for 7 min then gradient 70-95% A in 3min and hold at 95% A for 12 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
40 Isocratic 25% A for 25 min (20 mL/min flow rate). Mobile phase A was HPLC grade IPA, mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection method was UV, λ = 325 nm
41 Isocratic 10% A for 25 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection method was UV, λ = 320 nm
42 Isocratic 20% A for 20 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
43 Isocratic 15% A for 30 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IB, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
44 Isocratic 25% A for 25 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
45 The gradient was 10-60% A in 20 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1 % diethylamine added. The column used for the chromatography was a Daicel IC, 20 Method Conditions
x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
46 The gradient was 10-50% A in 13 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
47 The gradient was 10-50% A in 17 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
48 The gradient was 15-60% A in 17 min (20 mL/min flow rate). Mobile phase A was a 50:50 mixture of HPLC grade MeOH and EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1 % diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
49 Isocratic 25% A for 17 min then step to 60% A and hold for 10 min (20 mL/min flow rate). Mobile phase A was HPLC grade IP A, mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IC, 20 x 250 mm column (5 μιη particles). Detection method was UV, λ = 340 nm
50 Isocratic 20% A for 20 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.1% diethylamine added. The column used for the chromatography was a Daicel IB, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
51 Isocratic 10% A for 60 min (20 mL/min flow rate). Mobile phase A was a 50:50 mixture of HPLC grade MeOH and EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection Method Conditions
methods were evaporative light scattering (ELSD) detection as well as optical rotation.
52 Isocratic 50% A for 20 min (20 mL/min flow rate). Mobile phase A was a 50:50 mixture of HPLC grade MeOH and EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IC, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
53 The gradient was 30-70% A in 18 min with a hold at 70% for 4 min then re- equilibrate at 30% A for 13 min (20 mL/min flow rate). Mobile phase A was IP A, mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IC, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection as well as optical rotation.
54 Isocratic 30% A for 30 min (20 mL/min flow rate). Mobile phase A was ethanol (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IC, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
55 Isocratic 30% A for 30 min (20 mL/min flow rate). Mobile phase A was ethanol (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a (R,R) Whelk-Ol, 21 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
56 Isocratic 35% A for 30 min (20 mL/min flow rate). Mobile phase A was ethanol (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IC, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
57 Isocratic 30% A for 30 min (20 mL/min flow rate). Mobile phase A was a 50:50 mixture of HPLC grade MeOH and EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IC, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
Method Conditions
58 Isocratic 15% A for 1 1 min then step to 50% A in 0.5 min and hold for 4.5 min (20 mL/min flow rate). Mobile phase A was HPLC grade ethanol (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
59 The gradient was 10-95% A in 17 min with a hold at 95% for 2 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
60 Isocratic 20% A for 10 min then step to 60% A in 0.5 min and hold at 60% for 5.5 min (20 mL/min flow rate). Mobile phase A was HPLC grade IPA, mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
61 The gradient was 10-20% A in 28 min, hold at 20% for 2 min then 20-70% A in 5min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IC, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
62 Isocratic 22% A for 30 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IB, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
63 Isocratic 25% A for 30 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IB, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
64 Isocratic 65% A for 30 min (20 mL/min flow rate). Mobile phase A was ethanol Method Conditions
(200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a (R,R) Whelk-Ol, 21 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
65 Isocratic 65% A for 6 min then step to 90% A in 0.5 min and hold at 90% for 6.5 min (20 mL/min flow rate). Mobile phase A was ethanol (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a (R,R) Whelk-Ol, 21 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
66 Isocratic 30% A for 30 min (20 mL/min flow rate). Mobile phase A was HPLC grade IPA, mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IC, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
67 Isocratic 55% A for 8 min then step to 90% A in 1 min and hold at 90 for 7 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IC, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
68 The gradient was 60-90% A in 4 min with a hold at 90% for 6 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
69 Isocratic 20% A for 12 min then step to 50% A in 0.5 min and hold at 50% for 3.5 min (20 mL/min flow rate). Mobile phase A was EtOH (200 proof), mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IA, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
70 The gradient was 20-50% B over 10 min (0.6 mL/min flow rate). Mobile phase A was 10 mM KH2PO4 buffer (pH = 6.9) and mobile phase B was HPLC grade
Method Conditions
MeCN. The column used for the chromatography was a 4.6 x 150 mm Chiralpak AS-RH, Diacel col. Detection method was UV.
71 The gradient was 15-85% A in 37 min with a hold at 85% A for 0.5 min (20 mL/min flow rate). Mobile phase A was HPLC grade IP A, mobile phase B was HPLC grade heptane with 0.12% diethylamine added. The column used for the chromatography was a Daicel IC, 20 x 250 mm column (5 μιη particles). Detection methods were evaporative light scattering (ELSD) detection, and/or UV (variable wavelength) as well as optical rotation.
Purification Methods
For the general procedures, the intermediates and final compounds may be purified by any technique or combination of techniques known to one skilled in the art. Some examples that are not limiting include flash chromatography with a solid phase (e.g. silica gel, alumina, etc.) and a solvent (or combination of solvents) that elutes the desired compounds (e.g. heptane, EtOAc, DCM, MeOH, MeCN, water, etc.); preparatory TLC with a solid phase (e.g. silica gel, alumina etc.) and a solvent (or combination of solvents) that elutes the desired compounds (e.g. heptane, EtOAc, DCM, MeOH, MeCN, water, etc.); reverse phase HPLC (see Table 1 for some non-limiting conditions); recrystalization from an appropriate solvent or combination of solvents (e.g. MeOH, EtOH, IP A, EtOAc, toluene, etc.) or combination of solvents (e.g. EtOAc/heptane, EtOAc/MeOH, etc.); chiral LC with a solid phase and an appropriate solvent (see Table 2 for some non-limiting conditions)to elute the desired compound; chiral SFC with a solid phase and CO2 with an appropriate modifier (e.g. MeOH, EtOH, IPA with or without additional modifier such as diethylamine, TFA, etc.); precipitation from a combination of solvents (e.g. DMF/water, DMSO/DCM, EtOAc/heptane, etc.); trituration with an appropriate solvent (e.g. EtOAc, DCM, MeCN, MeOH, EtOH, IPA, «-ΙΡΑ, etc.); extractions by dissolving a compound in a liquid and washing with an appropriately immiscible liquid (e.g. DCM/water, EtOAc/water, DCM/saturated aqueous NaHC03, EtOAc/saturated aqueous NaHC03, DCM/10% aqueous HC1, EtOAc/10% aqueous HC1, etc.); distillation (e.g. simple, fractional, Kugelrohr, etc.); gas chromatography using an appropriate temperature, carrier gas and flow rate; sublimation at an appropriate temperature and pressure; filtration through a media (e.g. Florosil®, alumina, Celite®, silica gel, etc.) with a solvent (e.g. heptane, hexanes, EtOAc, DCM, MeOH, etc.) or combination of solvents; salt formation with solid support (resin based, e.g. ion exchange) or without. Compounds of interest may be isolated as a salt without the use of a specific salt formation purication method. For example, on occasions where purification is accomplished with reverse phase HPLC with an aqueous TFA buffer, the TFA salt may be isolated. Some descriptions of
these techniques can be found in the following references: Gordon, A. J. and Ford, R. A.. “The Chemist’s Companion”, 1972; Palleros, D. R. “Experimental Organic Chemistry”, 2000; Still, W. C, Kahn and M. Mitra, A. J. Org. Chem. 1978, 43, 2923; Yan, B. “Analysis and Purification Methods in Combinatorial Chemistry”, 2003; Harwood, L. M., Moody, C. J. and Percy, J. M. “Experimental Organic Chemistry: Standard and Microscale, 2nd Edition”, 1999; Stichlmair, J. G. and Fair, J. R. “Distillation; Principles and Practices”, 1998; Beesley, T. E. and Scott, R. P. W. “Chiral Chromatography”, 1999; Landgrebe, J. A. “Theory and Practice in the Organic Laboratory, 4th Ed.”, 1993; Skoog, D. A. and Leary, J. J. “Principles of Instrumental Analysis, 4th Ed.”, 1992; G. Subramanian, “Chiral Separation Techniques, 3rd Edition”, 2007; Y. Kazakevich, R. Lobrutto, “HPLC for Pharmaceutical Scientists”, 2007.