














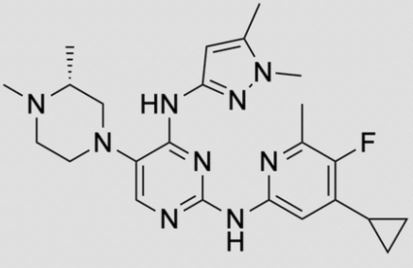

![2-N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-[(3R)-3,4-dimethylpiperazin-1-yl]-4-N-(1,5-dimethylpyrazol-3-yl)pyrimidine-2,4-diamine.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=92045019&t=l)
ZY 19489, MMV 253
C24 H32 FN9, 465.5
CAS 1821293-40-6
MMV253, GTPL10024, MMV674253
N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-((3R)-2-((1,5-dimethyl-1H-pyrazol-3-yl)amino)-3,4-dimethylpiperazin-1-yl)pyrimidin-2-amine
2-N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-[(3R)-3,4-dimethylpiperazin-1-yl]-4-N-(1,5-dimethylpyrazol-3-yl)pyrimidine-2,4-diamine
- N2-(4-Cyclopropyl-5-fluoro-6-methyl-2-pyridinyl)-5-[(3R)-3,4-dimethyl-1-piperazinyl]-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-2,4-pyrimidinediamine
- (R)-N2-(4-Cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine

SYN
IN 201721031453
The invention relates to triaminopyrimidine compd. of formula I, pharmaceutically acceptable salts thereof, hydrates, solvates, polymorphs, optically active forms thereof, in solid state forms useful for preventing or treating malaria. The invention also relates to a process for prepn. of triaminopyrimidine compd. and intermediates thereof. Compd. I was prepd. by condensation of 5-bromouracil with tert-Bu (R)-2-methylpiperazine-1-carboxylate to give tert-Bu (R)-4-(2,4-dichloropyrimidin-5-yl)-2-methylpiperazine-1-carboxylate, which underwent chlorination followed by condensation with 1,5-dimethyl-1H-pyrazol-3-amine followed by condensation with 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride to give (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine, which underwent Boc-deprotection followed by methylation to give I.
SYN
WO 2019049021
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019049021
Malaria is caused by protozoan parasites of the genus Plasmodium that infect and destroy red blood cells, leading to fever, severe anemia, cerebral malaria and, if untreated, death.
International (PCT) Publication No. WO 2015/165660 (the WO ‘660) discloses triaminopyrimidine compounds, intermediates, pharmaceutical compositions and methods for use for preventing or treating malaria. The WO ‘660 discloses a process for preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine (compound 5) as depicted in scheme-1.
Scheme 1
WO ‘660 discloses a process for preparation of triaminopyrimidine compounds depicted in scheme-2.
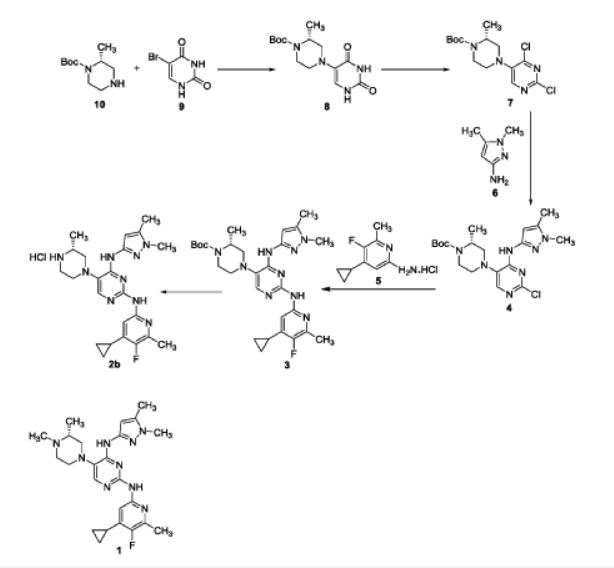
WO ‘660 discloses the preparation of compounds 8 and 4 by using microwave technique using Biotage microwave vial. WO ‘660 in example- 13, discloses the isolation of compound 1 by concentration of reaction mixture to obtain crude product, which was purified through reverse phase HPLC GILSON instrument to obtain pure solid compound 1 in 40.8% yield, without providing the purity of the solid compound 1. The process disclosed in WO ‘660 is not industrially advantageous as it requires microwave conditions as well as chromatographic purification and provides compound 1 with lower yields. The compound 1 prepared may not be suitable for pharmaceutical preparations based on various regulatory requirements.
Polymorphism, the occurrence of different crystalline forms, is a property of some molecules. A single molecule can exist in different crystalline forms having distinct physical properties like melting point, thermal behaviors (e.g. measured by thermogravimetric analysis – TGA, or different scanning calorimetry – DSC, Powder x-ray diffraction pattern – PXRD, infrared absorption – IR). One or more these techniques may be used to distinguish different polymorphic forms of a compound.
Different salts and solid states (e.g. solvates, hydrates) of an active pharmaceutical ingredient may possess different physio-chemical properties. Such variation in the properties of different salts and solid states forms may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, changing the dissolution profile in a favorable direction, or improving stability (both chemical and polymorph) and shelf-life. These variations in the properties of different salts and solid states forms may offer improvements to the final dosage form for example, to improve bioavailability. Different salts and solid state forms of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms or amorphous form, which may in turn provide additional opportunities to assess variations in the properties and characteristics of an active pharmaceutical ingredient.
In view of the above, the present invention provides a process for the preparation of triaminopyrimidine compound 1 or pharmaceutically acceptable salts thereof or hydrates or solvates or polymorphs or optically active forms thereof, which is industrially scalable, environment friendly and efficient so as to obtain compounds of the invention in higher yields and purity.
The process for the preparation of triaminopyrimidine compound 1 or intermediates thereof of the present invention, takes the advantage by using appropriate solvent systems and isolation techniques as well as purification techniques, thereby to overcome problems of lower yields, chromatography purifications and microwave reactions of the prior art.
SUMMARY OF THE INVENTION
The present invention provides solid state forms of triaminopyrimidine compound
1,
1
Examples: Preparation of Intermediates
Example-1: Preparation of 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine
In a 250 mL 4N round bottom flask, process water (30 ml) and cyclopropanecarboxylic acid (14.19 g, 164.88 mmol) were added at 25 to 35°C and started stirring. Sulphuric acid (4.4 ml, 82.44 mmol) was charged to the reaction mixture. Silver nitrate (4.18 g, 24.73 mmol), 6-Chloro-3-fluoro-2-methylpyridine (6 g, 41.22 mmol) were charged to the reaction mixture. Aqueous solution of ammonium persulphate (65.85 g, 288.54 mmol in 90 mL water) was added to the reaction mixture in 30 to 60 min at temperature NMT 60 °C. After the completion of the reaction as monitored by HPLC, toluene (30 ml) was added to the reaction mixture and stirred for 15 min. The reaction mixture filtered, separated layers from filtrate and extracted aqueous layer using toluene (30 mL). The organic layer was washed with aqueous sodium carbonate solution (30 mL) and water. The organic layer was distilled completely under vacuum at 60 °C to obtain 3.37 g syrupy mass as titled compound.
Example-2: Preparation of 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine
In a suitable glass assembly, process water (7.5 L) and cyclopropanecarboxylic acid (3.55 Kg, 41.24 mol) were added at 25 to 35 °C and stirred. Sulphuric acid (2.02 Kg, 20.59 mol), silver nitrate (1.05 Kg, 6.21 mol), 6-chloro-3-fluoro-2-methylpyridine (1.5 Kg, 10.3 mol) were added to the reaction mixture. Aqueous solution of ammonium persulphate (16.46 g, 72.13 mmol in 22.5 L water) was added to the reaction mixture at 55 to 60 °C and maintained. After the completion of the reaction as monitored by HPLC, toluene (7.5 L) was added to the reaction mixture and stirred for 15 min. The reaction mixture was filtered, organic layer was separated and aqueous layer was extracted using toluene (6 L), filtered the reaction mixture and washed the solid with toluene (1.5 L). The combined organic layer was washed with 20% sodium carbonate solution (9 L) and water. The organic layer was concentrated completely under vacuum at 60 °C to obtain 880 g (86.50%) syrupy mass of titled compound.
Example-3: Preparation of N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenyl-methanimine
In a 100 mL 3N round bottom flask, 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine (2.69 g, 14.48 mmol) and toluene (30 mL) were added at 25 to 35 °C. Diphenylmethanimine (3.15 g, 17.38 mmol) was charged to the reaction mixture and stirred for 5-10 min under nitrogen purging. Racemic BINAP (270 mg, 0.43 mmol) and palladium acetate (98 mg, 0.43 mmol) were added to the reaction mixture. Sodium-ie/ -butoxide (2.78 g, 28.96 mmol) was added to the reaction mixture and heated to 100 to 110° C under nitrogen. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C and filtered over hyflo bed and washed with toluene. The filtrate containing titled compound was preserved for next stage of reaction.
Example-4: Preparation of N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenyl-methanimine
In a suitable assembly, 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine (880) and toluene (7.5 L) were added at 25 to 35 °C. Diphenylmethanimine (787 g, 4.34 mmol) and BOC anhydride (237 g, 1.086 mol) was added to the reaction mixture and stirred for 5-10 min under nitrogen purging. Racemic BINAP (67.6 g, 0.108 mmol) and palladium acetate (24.4 g, 0.108 mol) were added to the reaction mixture. S odium- ieri-butoxide (870 g, 9.05 mol) was added to the reaction mixture and heated to 100 to 110 °C under nitrogen. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C, water (6 L) was added. The reaction mixture was filtered over hyflo bed and washed with toluene. The filtrate containing titled compound was preserved for next stage of reaction.
Example-5: Preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride monohydrate
In a 100 mL 3N round bottom flask, N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenylmethanimine in toluene as obtained in example-3 was added water (25 mL) at 25 to 35° C. The cone. HCl (3 mL) was charged to the reaction mixture and heated to 40 to 50 °C. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C. Layers were separated. The aqueous layer was treated with methylene dichloride and pH was adjusted to 7.5 to 8.5 using sodium carbonate solution, stirred for 15 min and layers were separated. Aqueous layer was extracted with methylene dichloride, charcoaled and acidified to pH 3 to 4 with aqueous HCl. The solvent was distilled completely and acetonitrile (9 mL) and ethyl acetate (9 mL) was added. The reaction mixture was stirred for 1 hour at 25 to 35° C. The product was filtered and washed with ethyl acetate. The product was dried at 50° C for 4 hours under vacuum to obtain 1.62 g title compound as monohydrate yellow crystalline solid having 99.51% HPLC purity.
Example-6: Preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride monohydrate
In a suitable glass assembly, N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenylmethanimine in toluene as obtained in example-4 was added water (6 L) at 25 to 35° C. The cone. HCl (750 mL) was charged to the reaction mixture and heated to 40 to 50 °C. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C. Layers were separated. The aqueous layer was treated with methylene dichloride (3 L) and pH was adjusted to 7.5 to 8.5 using sodium carbonate solution, stirred for 15 min and layers were separated. Aqueous layer was extracted with methylene dichloride (3 L), charcoaled and acidified to pH 3 to 4 with aqueous HCl. The solvent was distilled completely and acetonitrile (1.5 L) and ethyl acetate (1.5 L) were added. The reaction mixture was stirred for 1 hour at 25 to 35° C. The product was filtered and washed with ethyl acetate. The product was dried at 50° C for 4 hours under vacuum to obtain 489 g (96.80%) title compound as monohydrate yellow crystalline solid having 99.51% HPLC purity. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.5), Differential scanning calorimetry (FIG.6) and Thermogravimetric analysis (FIG.7).
Example 7: Preparation of 2,3-dibromobutanenitrile
In a 2 L round bottom flask, dichloromethane (550 mL) and 2-butenenitrile 110 g
(1.64 mol) were cooled to 20 to 25 °C. A solution of bromine 275 g (1.72 mol) in dichloromethane (220 mL) was dropwise added at 20 to 25 °C. Hydrobromic acid 1.43 ml (0.0082 mol) in acetic acid (33%) solution was added into the reaction mixture and stirred for 4 hours. After the completion of reaction, Na2S203 (550 mL) 4% aqueous solution was added and the reaction mixture was stirred for 15 min. The separated organic layer was distilled under vacuum completely to obtain 364.2 g (97.9%) of title compound as an oil.
Example 8: Preparation of l,5-dimethyl-lH-pyrazol-3-amine
In a 5 L round bottom flask, water (1. 36 L), sodium hydroxide 340 g (8.99 mol) were added and the reaction mixture was cooled to 0 to 5°C. A solution of methyl hydrazine sulphate 237.8 g (1.65 mol) in 680 mL water was added dropwise to the reaction mixture and stirred below 10 °C. 2,3-dibromobutanenitrile 340 g (1.5 mol) prepared in example-7 was added and the reaction mixture was stirred below 10 °C for 2 hours. After the completion of reaction, toluene (630 mL) was added and the reaction mixture was stirred for 15 min. The aqueous layer was separated and the organic layer was removed. The aqueous layer was extracted with dichloromethane (5.1 L). The combined organic layer was distilled completely under vacuum to obtain residue. Diisopropyl ether (680 mL) was added and the reaction mixture was stirred at 0 to 5 °C for 1 hour. The reaction mixture was filtered, washed with diisopropyl ether and dried to obtained 121.5 g (72.93%) of title compound having 95.63% purity.
Examples: Preparation of triaminopyrimidine compounds
Example-9: Preparation of tert-butyl (R)-4-(2,4-dioxo-l,2,3,4-tetrahydro- pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 2 L four neck round bottom flask, 1.25 Kg (6.545 mol) 5-bromouracil, 1.87 Kg (9.360 mol) tert-butyl (R)-2-methylpiperazine-l-carboxylate and 5L pyridine were added at 25 to 35° C. The reaction mass was stirred for 15 hours at 115 to 120°C. After completion, the reaction mass was cooled to 25 to 35°C. 12.5 L water was added and stirred for 1 hour. The reaction mass was filtered, washed with 2.5 L water and dried to obtain 1.37 Kg (67.4%) of title compound.
Example-10: Preparation of tert-butyl (R)-4-(2,4-dichloropyrimidin-5-yl)-2-methylpiperazine- 1 -carboxylate
In 20 L four neck round bottom flask, 1.36 Kg (4.382 mmol) tert-butyl (R)-4-(2,4-dioxo-1, 2,3, 4-tetrahydropyrimidin-5-yl)-2-methylpiperazine-l -carboxylate and 6.8 L phosphorus oxychloride were added at 25 to 35° C. 26.5 mL pyridine (0.329 mol) was added and the reaction mass was heated to 105 to 110 °C and stirred for 4 hours. After the completion of the reaction, phosphorus oxychloride was distilled completely at atmospheric pressure. 2.72 L acetone was added and the reaction mixture was quenched into 4.08 L water. Acetone was removed by distillation under vacuum. 20% sodium carbonate solution was added to adjust pH 7.5-8.5 of the reaction mixture. 1.14 Kg (5.258 mol) di-tert-butyl dicarbonate and 9.52 L ethyl acetate were added and stirred for 2 hours at 25 to 35 °C. After the completion of the reaction, the organic layer was separated and aqueous layer was extracted with 6.8 L ethyl acetate. The combined ethyl layers were distilled to remove ethyl acetate completely under vacuum to obtain residue. 1.36 L isopropyl alcohol was added to the residue and isopropyl alcohol was removed completely. 4.08 L isopropyl alcohol and 6.8 L water were added to the residue and stirred for 1 hour. The reaction mass was filtered, washed with water and dried to obtain 1.25 Kg of title compound.
Example-11: Preparation of tert-butyl (R)-4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate
In 20 L round bottom flask, 640 g (1.843 mol) tert-butyl (R)-4-(2, 4-dichloropyrimidin-5-yl)-2-methylpiperazine-l -carboxylate, 225.3 g (2.027 g) 1,5-dimethyl-lH-pyrazol-3-amine and 9.6L toluene were added at 25 to 35°C. 1.2 Kg (3.686 mol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. 12.41 g (0.0553 mol) palladium acetate and 34.43 g (0.0553 mol) racemic 2,2′-bis(diphenylphosphino)-l,l’-binaphthyl were added and the reaction mass was maintained for 16 hours at 110 to 115 °C under nitrogen. After the completion of the reaction, the reaction mixture was filtered through a celite bed and washed the bed with 1.28 L toluene. Toluene was distilled completely and 2.56 L dichlromethane was added. The compound was adsorbed by 1.92 Kg silica gel (60-120 mesh). The dichloromethane was distilled completely under vacuum and 12.8 L mixture of ethyl acetate and hexane was added to the residue and stirred for 2 hours. The silica gel was filtered and the filtrate was distilled completely under vacuum to obtain 595 g title compound.
Example-12: Preparation of tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 20 L round bottom flask, 595 g (1.40 mol) tert-butyl (R)- 4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate, 305 g (1.38 mol) 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride and 11.5 L toluene were added at 25 to 35°C. 1.08 Kg (3.32 mol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. 17.21 g (27.6 mmol) palladium acetate and 6.21 g (27.6 mmol) racemic 2,2′-bis(diphenylphosphino)-l, -binaphthyl were added. The reaction mass was stirred for 6 hours at 110 tol l5 °C under nitrogen. After the completion of the reaction, the reaction mixture was filtered through a celite bed and washed with toluene. The filtrate was used as such in the next step without further treatment.
Example-13: Preparation of tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 500 mL four neck round bottom flask, 7.5 g (17.77 mmol) (R)-tert-butyl 4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate, 3.92 g (17.77 mmol) 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride compound and 150 mL toluene were added at 25 to 35 °C. 20 g (61.3 mmol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. Then, 130 mg (0.58 mmol) palladium acetate and 360 mg (0.58 mmol) racemic 2,2′-bis(diphenylphosphino)-l,l’-binaphthyl were added. The reaction mass was stirred for 18 hours at 110 to 115° C under nitrogen. After completion, the reaction mixture was filtered through a celite bed and washed with toluene. The filtrate was used as such in the next step without further treatment.
2 4
Example-14: (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(1, 5-dimethyl-lH-pyrazol-3-yl)-5-(3-methylpiperazin-l-yl)pyrimidine-2,4-diamine
In 50 L glass assembly, the filtrate containing tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate from example 13 was taken. 11.5 L water and 1.28 L Cone. HC1 were added at 25 to 35 °C. The reaction mass was stirred for 2 hours at 50 to 55 °C. After the completion of the reaction, reaction mixture was cooled to room temperature and filtered over celite bed and washed with water. The separated the aqueous layer from filtrate was basified by using 20% sodium carbonate solution and extracted with 12.8 L methylene dichloride. The organic layer was distilled completely under vacuum to obtain residue. 9.6 L acetonitrile was added to the residue and heated to reflux for 30 min. The reaction mixture was cooled and stirred at 25 to 35 °C for 1 hour. The reaction mixture was filtered, washed with 640 mL acetonitrile and dried to obtain 360 g titled compound.
2 4
Example-15: (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(1,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine
In 250 mL four neck round bottom flask, 4.7 g (10.4 mmol) (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine was dissolved in 56 mL ethanol. 1.89 g (23.32 mmol) formaldehyde and 1.44 g (22.90 mmol) sodium cyanoborohydride were added. Adjusted pH 5-6 using acetic acid and stirred the reaction mass at 25 to 35 °C for 2 hours. After completion, ethanol was distilled completely under vacuum. 47 mL water was added to the residue. The reaction mass was basified by 20% sodium carbonate solution and extracted with methylene dichloride. Both the organic layers were combined and distilled completely under vacuum. 94 mL acetonitrile was added to the residue and heated to reflux for 15 min. The reaction mass was cooled to 25 to 35° C and stirred for 1 hour. The reaction mass was filtered, washed with 5 mL acetonitrile and dried to obtain 3.7 g title compound as crystalline solid, having HPLC purity of about 99.61%.
2 4
Example-16: (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(1,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine
In 20 L round bottom flask, 725 g (1.60 mol) (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazine-l-yl)pyrimidine-2,4-diamine was dissolved in 6.52 L dichloromethane. 261.5 g (3.2 mol) formaldehyde and 510.4 g (2.4 mol) sodium triacetoxyborohydride were added and stirred the reaction mixture at 25 to 35 °C for 2 hours. After the completion of the reaction, 3.63 L water was added into the reaction mixture. The reaction mixture was basified by 20% sodium carbonate solution and the organic layer was separated. The aqueous layer was extracted with 1.45 L methylene dichloride. The combined organic layers were distilled completely under vacuum. 14.5 L acetonitrile was added to the residue and heated to reflux for 15 min. The reaction mixture was cooled to 25 to 35° C and stirred for 1 hour. The reaction mass was filtered, washed with 1.45 L acetonitrile and dried to obtain 632 g of title compound as crystalline solid having 99.01% HPLC purity. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.l) and Differential Scanning Calorimetry (FIG.2).
2 4
Example-17: Preparation of (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine In a 10 mL round bottom flask, 300 mg (0.644 mmol) (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine, 2.7 mL acetonitrile and 0.3 mL water were added and the reaction mixture was heated to reflux for 15 min. The reaction mixture was cooled to 25 to 35 °C and stirred for 1 hour. The reaction mass was filtered, washed with acetonitrile and dried to obtain 201 mg (67%) title compound as crystalline solid. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.3) and Differential Scanning Calorimetry (FIG.4).
SYN
WO 2015165660
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015165660
Example 13
Synthetic scheme 1
Synthetic scheme 2
(R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine
In a 50 mL round-bottomed flask (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine hydrochloride (190 mg, 0.42 mmol, Example 2) was taken in DCM (2 mL) to give a yellow suspension. To this Hunig’s Base (0.184 mL, 1.05 mmol) was added and the suspension turned clear. After 10 minutes, it turned into a white suspension. After another 10 minutes, the mixture was concentrated to dryness. Resultant residue was dissolved in ethanol (absolute, 99.5%) (3 mL) and formaldehyde (0.042 mL, 0.63 mmol) was added and stirred for 10 minutes. White suspension slowly cleared to yellow solution. To this clear solution sodium cyanoborohydride (26.4 mg, 0.42 mmol) was added in one portion to get white suspension. After 30 minutes LCMS showed completion of reaction. The reaction mixture was concentrated and the crude was purified through reverse phase HPLC GILSON instrument to get the pure solid of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (80 mg, 40.8 %).1H NMR (300
MHz, DMSO-d6) δ ppm 0.67 – 0.78 (m, 2 H) 1.00 (d, J=6.22 Hz, 3 H) 1.02 – 1.08 (m, 2 H) 1.96 – 2.10 (m, 1 H) 2.23 (s, 7 H) 2.30 – 2.38 (m, 4 H) 2.73 – 2.96 (m, 4 H) 3.33 (s, 3 H) 6.83 (s, 1 H) 7.67 (d, J=5.09 Hz, 1 H) 8.00 (s, 1 H) 8.03 (s, 1 H) 9.26 (s,1 H) MS (ES+), (M+H)+ = 466.45 for C21H32FN9.
SYN
Nature Communications (2015), 6, 6715.
https://www.nature.com/articles/ncomms7715
Hameed P., S., Solapure, S., Patil, V. et al. Triaminopyrimidine is a fast-killing and long-acting antimalarial clinical candidate. Nat Commun 6, 6715 (2015). https://doi.org/10.1038/ncomms7715
The widespread emergence of Plasmodium falciparum (Pf) strains resistant to frontline agents has fuelled the search for fast-acting agents with novel mechanism of action. Here, we report the discovery and optimization of novel antimalarial compounds, the triaminopyrimidines (TAPs), which emerged from a phenotypic screen against the blood stages of Pf. The clinical candidate (compound 12) is efficacious in a mouse model of Pf malaria with an ED99 <30 mg kg−1 and displays good in vivo safety margins in guinea pigs and rats. With a predicted half-life of 36 h in humans, a single dose of 260 mg might be sufficient to maintain therapeutic blood concentration for 4–5 days. Whole-genome sequencing of resistant mutants implicates the vacuolar ATP synthase as a genetic determinant of resistance to TAPs. Our studies highlight the potential of TAPs for single-dose treatment of Pf malaria in combination with other agents in clinical development.

(A) Pyridine, microwave, 150 °C, 45 min. (B) (i) POCl3, reflux, 6 h (ii) sodium carbonate, di-tert-butyl dicarbonate, room temperature, 16 h. (C) N,N-Diisopropylethylamine (DIPEA), ethanol, microwave, 110 °C, 1 h. (D) (i) Potassium tert-butoxide, 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP), pd2(dba)3, toluene, reflux, 12 h. (E) HCl (4 N) in dioxane, 15–30 min. (F) Compound 9, DIPEA, dichloromethane, formaldehyde (HCHO), sodium cyanoborohydride, 15 min.
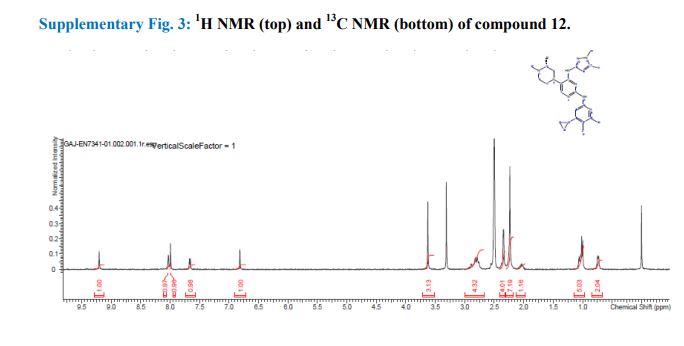
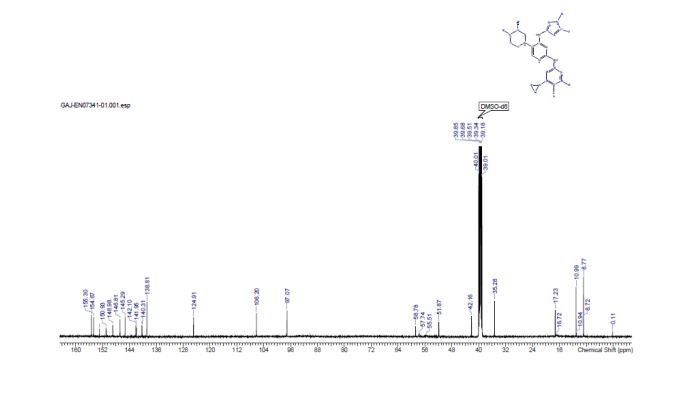
Synthesis of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1, 5-dimethyl-1H-pyrazol-3-yl)-5-(3, 4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (12). (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine hydrochloride (compound 9, 190 mg, 0.42 mmol) was taken in dichloromethane (2 ml) to give a yellow suspension. To this Hunig’s Base (0.184 ml, 1.05 mmol) was added and the suspension turned clear. After 10 min of stirring, reaction mixture turned into a white suspension and then it was concentrated to dryness. Resultant residue was dissolved in ethanol (absolute, 99.5%) (3 ml), and formaldehyde (0.042 ml, 0.63 mmol) was added and stirred for 10 min. To this clear solution, sodium cyanoborohydride (26.4 mg, 0.42 mmol) was added in one portion to get a white suspension. The reaction mixture was concentrated and the crude product was purified through reverse-phase chromatography to get the pure off-white solid of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1, 5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (80 mg, 40.8%). Yield: 40.8%, purity: >95% by HPLC (ultraviolet at 220 and 254 nm). 1H NMR (300 MHz, DMSO-d6) δ 9.26 (s,1H), 8.03 (s, 1H) 8.00 (s, 1H) 7.67 (d, J=5.1 Hz, 1H) 6.83 (s, 1H) 3.33 (s, 3H) 2.96–2.73 (m, 4H) 2.75–2.50 (m, 1H) 2.38–2.30 (m, 4H) 2.23 (s, 7H) 2.10–1.96 (m, 1H),1.08–1.02 (m, 2H) 1.00 (d, J=6.2 Hz, 3H) 0.78–0.67 (m, 2H). 13C-NMR (126 MHz, DMO-d6) δ 155.30, 154.67, 152.10, 150.93, 148.98, 146.81. 145.29, 141.95, 140.31, 138.81, 124.91, 106.20, 97.07, 58.78, 51.87, 42.16, 35.28, 17.23. 10.99 and 8.77, HRMS (ESI): m/z calculated for C24H32FN9+H [M+H]: 466.2765. Found: 466. 2838. Traces of LC-MS, HRMS, 1H NMR and 13C-NMR of compound 12 are shown in Supplementary Figs 1–3.
| Product vision |
|
| MoA |
|
| Key features |
|
| Challenges |
|
| Status |
|
| Next milestone |
|
| Previously |
|
Zydus receives Orphan Drug Designation from USFDA for ZY-19489, a novel compound to treat malaria;
ZY19489 is a novel antimalarial compound active against all current clinical strains of P. falciparum and P. vivax, including drug-resistant strains.
Zydus Cadila listed as Cadila Healthcare Limited announced that its antimalarial compound ZY19489 (MMV253), currently in development together with Medicines for Malaria Venture (MMV), a leading product development partnership (PDP) in antimalarial drug research, has received Orphan Drug Designation from the USFDA.
Orphan drug designation provides eligibility for certain development incentives, including tax credits for qualified clinical testing, prescription drug user fee exemptions, and seven-year marketing exclusivity upon FDA approval.
The company said that the Phase I study of ZY19489 has demonstrated a long half-life and potential for a single-dose cure for malaria. In a separate malaria challenge trial, potent antimalarial activity has been demonstrated following single-dose oral administration of ZY19489.
“As a global community facing threats from rapidly mutating malaria strains and the rise in artemisinin resistance cases, we have to be prepared with novel therapeutic drugs. ZY-19489 is a potential single dose radical cure for P. falciparum and P. vivax malaria which is a major global health risk today,” Pankaj R. Patel, Chairman, Zydus Group, said.
“ZY19489 is a potent, first in class molecule, originally discovered and elaborated in India” said Dr. Timothy Wells, Chief Scientific Officer, MMV. “It has tremendous potential as part of a new generation of treatments and is fully active against drug resistant strains of malaria which are increasingly a concern.”
Artemisinin resistance is seen as a mounting challenge to the global fight against malaria. ZY19489 is being developed to provide an effective alternative to the current front-line antimalarial drugs for the treatment of P. falciparum and P. vivax malaria, as artemisinin-based combination therapies (ACTs) are under threat of resistance.
As per the World Malaria Report 2021, there were an estimated 241 million cases of malaria worldwide and the estimated number of malaria deaths stood at 627,000 in 2020. A major health concern, it is estimated that a child dies from malaria every minute. About 96% of malaria deaths globally were in 29 countries. India accounted for about 82% of all malaria deaths in the WHO South-East Asia Region.
////////////ZY 19489, MMV 253, Orphan Drug Designation, PHASE 1, ZYDUS CADILA, ANTIMALARIAL
Cn1nc(Nc2nc(Nc3cc(C4CC4)c(F)c(C)n3)ncc2N2C[C@@H](C)N(C)CC2)cc1C
CC1CN(CCN1C)C2=CN=C(N=C2NC3=NN(C(=C3)C)C)NC4=NC(=C(C(=C4)C5CC5)F)C