














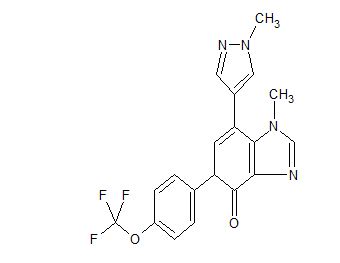
AK-3280
AK 3280; GDC3280; RG 6069
Ci8Hi4N502F3, mass 389.3 g/mol),
ROCHE,
Ark Biosciences , under license from Roche , is developing AK-3280, an antifibrotic agent, for the potential oral treatment of IPF. In July 2018, Ark intended to further clinical development of the drug, for IPF. In June 2019, a phase I trial was planned in Sweden.
- Originator Genentech
- Mechanism of Action Undefined mechanism
- Phase I Interstitial lung diseases
- 19 Jun 2019Ark Biosciences plans a phase I trial for Idiopathic pulmonary fibrosis (In volunteers) in Sweden (PO, Tablet), in August 2019 , (NCT03990688)
- 28 Sep 2018GDC 3280 is still in phase I trials for Interstitial lung diseases (Genentech pipeline, September 2018)
- 28 Jun 2018No recent reports of development identified for phase-I development in Fibrosis(In volunteers) in United Kingdom (PO)
Introduction
GDC 3280 (also known as RG 6069), an orally administered drug, is being developed by Genentech, for the treatment of interstitial lung diseases. Early stage clinical development is underway in the UK.
Company Agreements
In September 2018, Genentech licensed exclusive worldwide development and commercialisation rights of GDC 3280 to Ark Biosciences, for the treatment of idiopathic pulmonary fibrosis
Key Development Milestones
As at September 2018, GDC 3280 is still in phase I development for interstitial lung disease (Genentech pipeline, September 2018).
In December 2015, Genentech completed a phase I trial that evaluated the safety, pharmacokinetics and tolerability of GDC 3280 in healthy volunteers, compared with placebo (GB29751; EudraCT2015-000560-33; NCT02471859). The randomised, double-blind, single and multiple oral dose trial was initiated in June 2015 and enrolled eight volunteers in the UK .
PATENT
WO-2019152863
Novel crystalline salt forms of 1-methyl-7-(1-methyl-lH-pyrazol-4-yl)-5-(4-(trifluoromethoxy)phenyl)-1,5-dihydro-4H-imidazo[4,5-c]pyridin-4-one (compound I; presumed to be AK-3280 ), processes for their preparation and compositions comprising them are claimed.
Compound I is an orally available small molecule having the structure:
[0004] Compound I has therapeutic value in several different indications that display fibrotic pathophysiology, including idiopathic pulmonary fibrosis (IPF).
[0005] Idiopathic pulmonary fibrosis is a disease of unknown etiology that occurs mainly in middle-aged and elderly patients, which is characterized by progressive fibrosis of the lung, leading to pulmonary insufficiency and death. Because fibrosis has long been considered to be a clinically irreversible process, treatments have traditionally been focused on managing the symptoms and complications, with little hope of significantly slowing progression of the condition. For many years, mainstay treatments have been typically anti inflammatory, immunosuppressive, and anti-oxidant agents. The effectiveness of these therapies in the treatment of IPF and other fibrotic conditions appears to be minimal and variable, and their side effects are often poorly tolerated by patients.
[0006] New treatment options have only recently become available. Both pirfenidone and nintedanib have been approved for use in the treatment of IPF. Current research efforts to develop new anti-fibrotic agents are targeting multiple mechanisms proposed to be linked to the underlying molecular pathogenic processes. This changing landscape has raised hopes and expectations for what might be achievable with new single agents or combination therapies targeting additional pathways.
Preparation of Compound I and its salts
[0045] A synthesis of Compound I and its tosylate salt is shown in the scheme below:
[0046] l-methyl-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-4H-imidazo[4,5-c]pyridin-4-one (5) was synthesized in 4 steps, including a copper-catalyzed coupling reaction e.g., a Goldberg-Ullmann coupling reaction. In another aspect of the invention, intermediate (5) is synthesized using any transition metal-catalyzed coupling reaction. The skilled chemist would know that intermediate (5) could be synthesized from intermediate (4) and compounds
LG
of the general formula: OCF3 , wherein the leaving group“LG” includes but is not limited to halogen, tosylate, mesylate, triflate, etc.
[0047] Compound I was synthesized in 6 steps, using a transition metal cross-coupling reaction, e.g., a Suzuki reaction. In another aspect of the invention, Compound I is synthesized using any cross -coupling reaction. Compound I is synthesized from intermediate 6 containing any leaving group. For example, the skilled chemist would use compounds of
the general formula:
, wherein the leaving group“LG” includes but is not limited to halogen, tosylate, mesylate, triflate, etc.
An alternative synthesis of Compound I and its salts is shown in the scheme below:
Example 13 – Synthesis of Compound I Tosylate Salt
[00183] A process for the formation of mono- and di-tosylate salts of Compound I was developed and a batch was performed to successfully produce the mono-tosylate salt.
Step 1 : Synthesis of2-chloro-N-methyl-3-nitropyridin-4-amine
[00184] A reactor was charged with 2,4-dichloro-3-nitropyridine and 3.0 volumes of DMF. The solution was stirred at 20-25 °C until a clear solution was obtained. The solution was then cooled to 0-5 °C, and 2.1 equivalents of 40% methylamine in water were slowly added over at least 2 hours at 0-5 °C. The reaction mixture was stirred for at least 2 hours at 0-5 °C until conversion to the product was 95% (as measured by HPLC). The reaction mixture was diluted by slowly adding 10 volumes of water over at least 30 minutes at 0-5 °C. The obtained suspension was stirred for at least 60 minutes at 0-5 °C. The precipitate was collected by filtration, and the filter cake was rinsed via the reactor with 10 volumes of water at 0-5 °C. The damp filter cake was then dried in a flow of dry nitrogen to yield 2-chloro-A-methyl-3-nitropyridin-4-amine in 78% yield.
Step 2: Synthesis of 2-chloro-N4 -methylpyridine-3, 4-diamine
[00185] A reactor was charged with catalyst [2% Pt on charcoal, 59 %wt. water] (0.0004 equivalents Pt), damp 2-chloro-/V-methyl-3-nitropyridin-4-amine from step 1 and 9.4 volumes of THF. The solution was stirred, and then the suspension was transferred from the glass-reactor to an autoclave. The line was rinsed with 1.2 volumes of THF into the autoclave, and the autoclave was purged with nitrogen for 15 minutes at 50 rpm, followed by hydrogen for 15 minutes at 150 rpm. The autoclave was closed, and the hydrogen pressure was adjusted to 2 bar at 20-30 °C. The reaction mixture was stirred for 4-8 hours at 2 bar and 20-30 °C.
[00186] Next, the autoclave was released to atmospheric pressure and purged with nitrogen for at least 15 minutes. Conversion to the product was verified by HPLC, and then the catalyst was removed by filtration. The filtered catalyst was rinsed with 1.3 volumes of THF and the filtrates were combined. The combined filtrates were charged to a second reactor via a particle filter, and the line was rinsed with 0.5 volumes of THF. The solution was concentrated to a final volume of 2.5 volumes by distillation under reduced pressure at 40-45 °C.
[00187] The solution was then diluted with 10 volumes of THF in portions while concentrating the solution to a final volume of 2.5 volumes by distillation under reduced pressure at 45-50 °C. The reactor was purged with nitrogen to atmospheric pressure, and 5.0 volumes of heptane were added to the residue at 40-50 °C. The reaction mixture was cooled over 2 hours to 20-25 °C, and stirring was continued for 1 hour. The reaction mixture was then further cooled to 0-5 °C over 1 hour, and stirring was continued for 1 hour. The precipitated product was collected by filtration, rinsed via the reactor with 5.0 volumes of heptane, and the damp filter cake was dried in a vacuum drying oven at max. 40 °C until loss on drying was < 2 % weight, giving 2-chloro-/V4-methylpyridine-3, 4-diamine in 85% yield.
Step 3 : Synthesis of -inelhyl- 1 ,5-dihvdro-4H-iinidazoi4,5-c h yridin-4-one
[00188] A reactor was charged with 2-chloro-/V4-methylpyridine-3, 4-diamine and 4 volumes of formic acid. The reaction mixture was heated to smooth reflux within one hour, and reflux was maintained for 6 hours. The reaction mixture was then cooled to
approximately 60 °C, and conversion to the product was verified by HPLC.
[00189] The reaction mixture was then concentrated by distillation under reduced pressure at 60-80 °C to a final volume of 2 volumes. The temperature of the solution was adjusted to 60 °C, maintaining the temperature above 50 °C to avoid precipitation.
[00190] Next, a second reactor was charged with 10 volumes of acetone, and heated to gentle reflux. The product solution from the first reactor was slowly transferred to the acetone in the second reactor over 20 minutes, and the line was rinsed with approximately 0.05 volumes of formic acid. Reflux of the obtained suspension was maintained for 15 minutes. The slurry was cooled to 0 °C within 1 hour, and stirring was continued for 1 hour at that temperature. The precipitate was collected by filtration, and the filter cake was rinsed via the reactor with 3.7 volumes of cold acetone at 0-10 °C. The filter cake was dried in a flow of dry nitrogen or in a vacuum drying oven at 50 °C until loss on drying was < 2% of weight, giving 1 -methyl- 1 ,5-dihydiO-4/7-imidazo[4,5-c]pyndin-4-onc in 95% yield.
Step 4: Synthesis of l-methyl-5-(4-(trifluoromethoxy)phenyl)-J5-dihvdro-4H-imidaz.o[4,5-c]pyridin-4-one
[00191] A first reactor (Reactor A) was charged with 1 -methyl- 1 ,5-dihydro-4/7-imidazo[4,5-c]pyridin-4-one (1.0 mol equivalent), Cu(0Ac)2 H20 (0.1 mol equivalents), and K2C03 (1.1 mol equivalents). The reactor was closed and the atmosphere replaced with nitrogen.
[00192] Next, l-bromo-4-(trifluoromethoxy)benzene (1.5 mol equivalents) and N-methylpyrrolidinone (5.4 volume equivalents) were added, whereupon a suspension was formed. The suspension was stirred until the temperature had fallen again to approximately 20-25 °C and gas evolution had slowed. The reaction mixture was heated to approximately 130-150 °C at which time a blue/green color was observed, changing to dark brown after some time. The reaction was stirred at 130-150 °C for at least 40 hours. Stirring times of 40 hours up to 72 hours were required to reach an acceptable level of conversion. In general, higher reaction temperatures supported faster conversion.
[00193] Next, the reaction mixture was cooled to approximately 20-30 °C, and 25% aqueous NH3 (0.7 volume equivalents) was added, followed by water (3.5 volume equivalents). The resulting suspension was transferred into a second reactor (Reactor B). Additional water was added (18.1 volume equivalents) to the reaction mixture via Reactor A, followed by n-heptane (3.2 volume equivalents). The resulting suspension was cooled to approximately 0-5 °C, and stirred for approximately 2 hours.
[00194] The suspension was filtered, and the filter cake was washed with water (9.7 volume equivalents). The filter cake was then dissolved in dichloromethane (14.1 volume equivalents) and transferred back into reactor B. To this solution was added water (5.7 volume equivalents) via the filter, followed by 25% aq. NH3(1.6 volume equivalents). The mixture was stirred for approximately 1 hour at approximately 15-25 °C.
[00195] Next, the layers were separated, and dichloromethane was added (3.6 volume equivalents) to the aqueous layer. The biphasic mixture was stirred at approximately 15-25 °C for approximately 20-30 minutes. The layers were separated over a period of at least 1 hour, and to the combined organic layers was added a solution of NH4Cl (2.5 mol equivalents) in water (7.0 volume equivalents). The biphasic mixture was stirred at approximately 15-25 °C for about 20-30 minutes, then the layers were separated over the course of 1 hour.
[00196] The lower organic layer was filtered through a particle filter and diluted with toluene (7.1 volume equivalents) via the filter. The organic layer was concentrated under ambient pressure at approximately 80 °C, until no further liquid was seen to evaporate and a precipitate began to form. Toluene was added (16.6 volume equivalents), then concentrated in vacuo, followed by addition of more toluene (7.1 volume equivalents) and again concentrated in vacuo. The suspension was cooled to approximately 0-5 °C, stirred for approximately 2 hours, and filtered. The filter cake was washed with toluene (2.9 volume equivalents), and dried in vacuo at approximately 50 °C until the loss on drying was 0.5% of the weight to give l-methyl-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-47/-imidazo[4,5-c]pyridin-4-one as a beige-colored solid in 83.1% yield.
Step 5 : Synthesis of 7-bromo- 1 -methyl-5-(4-( trifluoromethoxy Iphenyl )- l,5- 4H-
imidaz.o[4,5-clpyridin-4-one
[00197] A first reactor (Reactor A) was charged with water (1.8 volume equivalents) and cooled to approximately 0-5 °C, to which was slowly added 96% sulfuric acid (14 mol. equivalents) at approximately 0-20 °C. The temperature of the solution was adjusted to approximately 0-5 °C, and l -mcthyl-5-(4-(tnfluoromcthoxy)phcnyl)-l ,5-dihydro-4/7-imidazo[4,5-c]pyridin-4-one (1.0 mol equivalent) was added in 3-4 portions at approximately 0-5 °C. The temperature of the mixture was adjusted to approximately 0-5 °C, and N-bromosuccinimide (1.0 mol equivalents) was slowly added in 3-4 portions, while maintaining the temperature at approximately 0-5 °C.
[00198] The reaction mixture was stirred for about 1 hour at approximately 0-5 °C, and then for an additional 4-16 hours at approximately 0-22 °C. Conversion to the product was confirmed by HPLC, then the reaction mixture was cooled to approximately 0-5 °C.
[00199] A second reactor (Reactor B) was charged with water (42.7 volume equivalents) and cooled to approximately 0-5 °C. The reaction mixture from Reactor A was transferred into the pre-cooled water in Reactor B at a temperature below 30 °C over 2 hours. The reaction was rinsed with water (1.6 volume equivalents), and 50% aqueous sodium hydroxide (25 mol. equivalents) was carefully added at approximately 0-30 °C over about 2 hours until the pH reached 2-5.
[00200] Next, MTBE (6.5 volume equivalents) was added at approximately 0-20 °C, and the mixture was stirred for about 5 minutes. Additional 50% aqueous sodium hydroxide (2 mol. equivalents) was added at approximately 0-30 °C until the pH of the solution was in the range of 10-14. The reaction was stirred for at least 1.5 hours at approximately 15-25 °C, and then the layers were allowed to separate over a period of at least 1 hour. The suspension was filtered, taking care to capture the product, which accumulated at the interface of the aqueous and organic layers. The filter cake was washed with MTBE (1.7 volume equivalents), water (3.0 volume equivalents), and then MTBE again (3.0 volume equivalents). The product was dried in vacuo at below 50 °C until the loss on drying was < 1% of the weight, giving 7-bromo-l-methyl-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-47/-imidazo[4,5-c]pyridin-4-one as a pale beige-colored solid in 97.6% yield.
Step 6: Synthesis of 1 -methyl-7 -( 1 -methyl-lH-pyraz.ol-4-yl )-5-(4-( trifluoromethoxy )pheml )-J5-dihvdro-4H-imidaz.o[4,5-c]pyridin-4-one (Compound /)
[00201] A reactor was charged with 7-bromo-l-methyl-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-4//-imidazo[4,5-c]pyridin-4-one (1.0 mol equivalents), ( 1 -methyl- 1 //-pyrazol-4-yl)boronic acid pinacol ester (l-methyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-l//-pyrazole, 1.6 mol equivalents), Pd[Ph3]4 (0.025 mol equivalents, and K2C03 (2.0 mol equivalents), to which were added acetonitrile (10.0 volume equivalents) and water (3.0 volume equivalents). The reaction mixture was stirred for approximately 10-20 minutes at about 20-25 °C to form a suspension.
[00202] The mixture was heated to slight reflux, whereupon a biphasic, yellow solution formed. The mixture was stirred at slight reflux for at least 10 hours. The reaction mixture was cooled to between 30-50 °C, then passed through a particle filter. The filter was washed with acetonitrile (2.6 volume equivalents), the filtrates were combined, and the solution was concentrated to a final volume of approximately 120 mL (4.8 volume equivalents) under reduced pressure at below 60 °C.
[00203] To the resulting suspension was added water (1.9 volume equivalents), methanol (26 mL, 1.0 volume equivalents), and dichloromethane (14.8 volume equivalents). The mixture was warmed to about 30-35 °C and stirred until two clear layers were observed. The layers were allowed to separate without stirring at about 30-35 °C, and additional dichloromethane (3.7 volume equivalents) was added to the aqueous layer. The mixture was warmed to approximately 30-35 °C and stirred for about 5 minutes, and then the layers were allowed to separate at approximately 30-35 °C.
[00204] To the combined organic layers was added water (1.9 volume equivalents), and the mixture was warmed to approximately 30-35 °C and stirred for about 5 minutes. The layers were separated at approximately 30-35 °C. Charcoal was added to the combined organic layers and stirred for 30-60 minutes at approximately 30-35 °C. The charcoal was removed by filtration, and the filter was washed with dichloromethane (39 mL, 1.6 volume equivalents).
[00205] The solution was concentrated to approximately 4.0 volume equivalents at ambient pressure and at below 50 °C, then diluted with methanol (5.0 volume equivalents). The solution was again concentrated to approximately 4.0 volume equivalents at ambient pressure and below 60 °C, diluted with methanol (5.0 volume equivalents), and concentrated to a final volume of approximately 3.0 volume equivalents under reduced pressure below 60 °C.
[00206] To the resulting suspension was added methanol (2.9 volume equivalents), and the suspension was warmed to approximately 45-55 °C and stirred for about 1 hour. The suspension was cooled to approximately 0-5 °C within approximately 1 hour, stirred for 1 hour at approximately 0-5 °C, and then filtered. The filter cake was washed with cold methanol (pre-cooled to approximately 0-10 °C, 2.9 volume equivalents), and the product was dried under a stream of nitrogen and in vacuo at below 60 °C until the loss on drying was < 1% by weight, giving Compound I (l-methyl-7-(l-methyl-l -pyrazol-4-yl)-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-4//-imidazo[4,5-c]pyridin-4-one) as a white solid in 88.5% yield.
Step 7: Recrystallization of 1 -methyl-7 -(1 -methyl- lH-pyraz.ol-4-yl)-5-( 4-(trifluoromethoxy)phenyl)-J5-dihvdro-4H-imidaz.o[4,5-c]pyridin-4-one (Compound /)
[00207] A reactor was charged with crude l-methyl-7-(l -methyl- l//-pyrazol-4-yl)-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-47/-imidazo[4,5-c]pyridin-4-one from step 6, and to this was added glacial acetic acid (1.5 volume equivalents). The suspension was warmed to approximately 50-60 °C and stirred until a clear solution was obtained, approximately 10-20 minutes. The warm solution was passed through a particle filter into a second reactor.
[00208] To this solution was added ethanol (10.0 volume equivalents) at approximately 45-55 °C over 2 hours. The suspension was stirred for approximately 30 minutes at approximately 45-55 °C, then cooled to approximately 0-5 °C over about 4 hours. The suspension was then stirred for approximately 4-16 hours at about 0-5 °C.
[00209] Next, the suspension was filtered and the filter cake was washed with cold isopropanol (4.2 volume equivalents) at approximately 0-20 °C. The product was dried under a nitrogen stream and in vacuo at below 60 °C until the loss on drying was < 1% by weight, giving Compound I ( 1 – mcthyl-7-( 1 -methyl- 1 /7-pyrazol-4-yl)-5-(4-(tnfluoromcthoxy)phcnyl)-l,5-dihydro-47/-imidazo[4,5-c]pyridin-4-one) as a white solid in 93.0% yield.
Step 8 : Synthesis of 1 -methyl-7 -( 1 -methyl- 1 H-pyrazol-4-yl )-5-(4-( trifluoromethoxy )phenyl )- 1 ,5-dihvdro-4H-imidaz.oi 4,5-clpyridin-4-one, mono – mono -tosylate
salt)
[00210] A reactor was charged with Compound I ( 1 -mcthyl-7-( 1 -methyl- 1 /7-pyrazol-4-yl)-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-4//-imidazo[4,5-c]pyridin-4-one, 1.00 mol equivalent), para-toluenesulfonic acid monohydrate (1.05 mol equivalents), acetone (6.75 volume equivalents), and water (0.75 volume equivalents). The mixture was stirred at 15-25 °C until a clear solution formed, and then this solution was filtered through a particle filter into a second reactor.
[00211] The filter was washed with acetone (2.5 volume equivalents), and to the combined filtrates was added MTBE (7.5 volume equivalents) at 15-25 °C and Compound I mono-tosylate seeding crystals (0.001 mol equivalents).
[00212] The resulting suspension was stirred at 15-25 °C for approximately 30-60 minutes, and MTBE was added (22.5 volume equivalents) at 15-25 °C during a period of
approximately 30 minutes. Stirring was continued at 15-25 °C for approximately 30-60 minutes, and then the suspension was filtered. The filter was washed with MTBE (2.5 volume equivalents), and the material was dried in vacuo at below 55 °C to give Compound I mono-tosylate salt (l-methyl-7-(l-methyl-l//-pyrazol-4-yl)-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-47/-imidazo[4,5-c]pyridin-4-one, mono-tosylate salt) as a white, crystalline solid in 93% yield.
PATENT
WO2018102323 ,
claiming use of a specific compound, orally administered, in combination with food (eg low, medium or high fat meal) for treating fibrotic, inflammatory or autoimmune disorders eg idiopathic pulmonary fibrosis IPF, assigned to Genentech Inc ,
References
-
Roche licenses IPF candidate to Ark Biosciences. Internet-Doc 2019;.
Available from: URL: https://scrip.pharmaintelligence.informa.com/deals/201820364
-
Roche Q3 2018. Internet-Doc 2018;.
Available from: URL: https://www.roche.com/dam/jcr:f9cad8fc-8655-4692-9a85-efbe1cf7a59b/en/irp181017.pdf
-
A Phase 1, Randomized, Double-Blind, Placebo-Controlled, Ascending, Single- and Multiple-Oral-Dose, Safety, Tolerability, and Pharmacokinetic Study of GDC-3280 in Healthy Subjects
// AK-3280, AK 3280, AK3280, GDC 3280, RG 6069, PHASE 1, Idiopathic pulmonary fibrosis