
















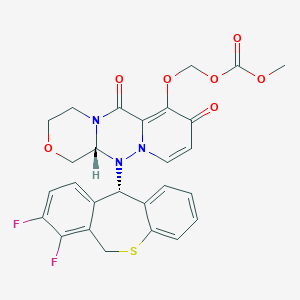
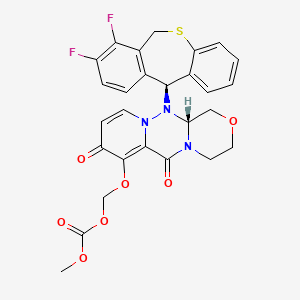
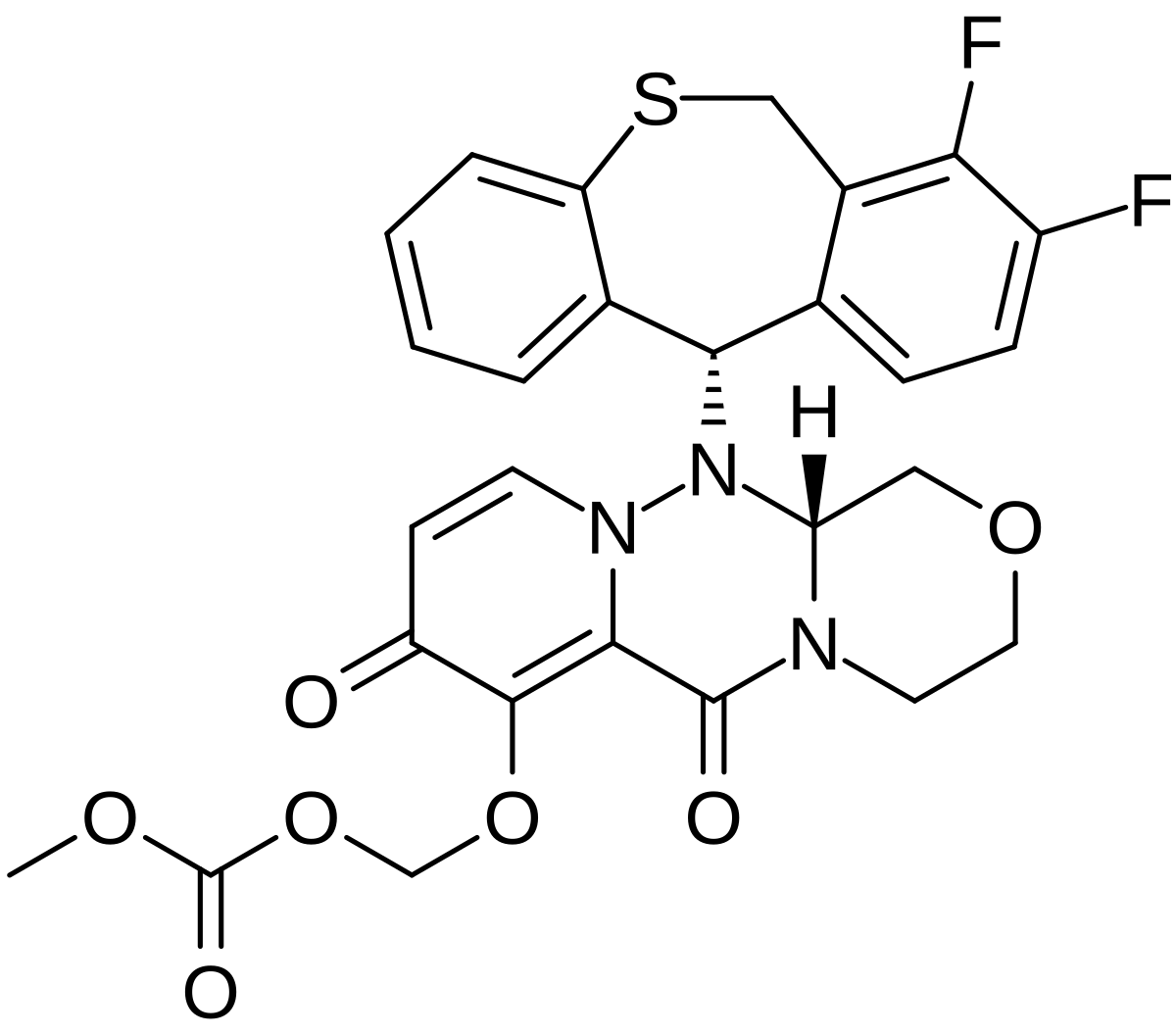
Baloxavir marboxil
バロキサビルマルボキシル
балоксавир марбоксил [Russian] [INN]
Carbonic acid, [[(12aR)-12-[(11S)-7,8-difluoro-6,11-dihydrodibenzo[b,e]thiepin-11-yl]-3,4,6,8,12,12a-hexahydro-6,8-dioxo-1H-[1,4]oxazino[3,4-c]pyrido[2,1-f][1,2,4]triazin-7-yl]oxy]methyl methyl ester
({(12aR)-12-[(11S)-
- (((12aR)-12-((11S)-7,8-Difluoro-6,11-dihydrodibenzo(b,E)thiepin-11-yl)-6,8-dioxo-3,4,6,8,12,12ahexahydro-1H-(1,4)oxazino(3,4-C)pyrido(2,1-F)(1,2,4)triazin-7-yl)oxy)methyl methyl carbonate
- Carbonic acid, (((12aR)-12-((11S)-7,8-difluoro-6,11-dihydrodibenzo(b,E)thiepin-11-yl)-3,4,6,8,12,12a-hexahydro-6,8-dioxo-1H-(1,4)oxazino(3,4-C)pyrido(2,1-F)(1,2,4)triazin-7-yl)oxy)methyl methyl ester
Antiviral
In Japan the product is indicated for treatment influenza types A and B in adults and children
RG-6152
- Originator Shionogi
- Developer Roche; Shionogi
- Class Antivirals; Dibenzothiepins; Esters; Pyridines; Small molecules; Triazines
- Mechanism of Action Endonuclease inhibitors
Highest Development Phases
- Marketed Influenza A virus infections; Influenza B virus infections
- Phase III Influenza virus infections
- Preclinical Influenza A virus H5N1 subtype
|
Xofluza (TN)
Antiviral
|
|
| Formula |
C27H23F2N3O7S
|
|---|---|
| Cas |
1985606-14-1
|
| Mol weight |
571.5492
|
| 2018/2/23 | PMDA | JAPAN | APPROVED | Baloxavir marboxil | Xofluza | Shionogi |

| バロキサビル マルボキシル Baloxavir Marboxil  C27H23F2N3O7S : 571.55 [1985606-14-1] |
![]()

https://chem.nlm.nih.gov/chemidplus/sid/1985606141
Baloxavir marboxil (trade name Xofluza, compound code S-033188/S-033447) is a medication being developed by Shionogi Co., a Japanese pharmaceutical company, for treatment of influenza A and influenza B. The drug was in late-stage trials in Japan and the United States as of early 2018, with collaboration from Roche AG.[1].
It was approved for sale in Japan on February 23, 2018.[2]
Baloxavir marboxil is a medication developed by Shionogi Co., a Japanese pharmaceutical company, for treatment of influenza A and influenza B. The drug was approved for use in Japan in February 2018 and is in late phase trials in the United States as of early 2018. Roche, which makes Tamiflu, has acquired the license to sell Xofluza internationally, but it may not be until 2019 that it could be available in the United States [7]. Interestingly, a study has determined that administering Baloxavir marboxil with neuraminidase inhibitors leads to a synergistic effect in influenza treatment

It is an influenza therapeutic agent (cap-dependent endonuclease inhibitor), characterized by only taking one dose. Unlike neuraminidase inhibitors such as oseltamivir (Tamiflu) and zanamivir (Relenza) that inhibit the action of neuraminidase, which liberates viruses from the infected cells surface, baloxavir marboxil may prevent replication by inhibiting the cap-dependent endonuclease activity of the viral polymerase.[3]
In October 2015, the Japanese Ministry of Health, Labour and Welfare granted Sakigake status to Shionogi’s baloxavir marboxil for A type or B -type influenza virus infection . In October 2015, the drug was designated for Priority Review by the Ministry of Health, Labour and Welfare, presumably for the treatment of A type or B -type influenza virus infection .
This drug is a CAP endonuclease inhibitor [1]. The influenza endonuclease is an essential subdomain of the viral RNA polymerase enzyme. CAP endonuclease processes host pre-mRNAs to serve as primers for viral mRNA and therefore has been a common target for studies of anti-influenza drugs.
Viral gene transcription is primed by short-capped oligonucleotides that are cleaved from host cell pre mRNA by endonuclease activity. Translation of viral mRNAs by the host ribosome requires that they are capped at the 5′ end, and this is achieved in cells infected with influenza virus by a “cap-snatching” mechanism, whereby the endonuclease cleaves 5′ caps from host mRNA which then act as primers for transcription.The N-terminal domain of PA subunit (PAN) has been confirmed to accommodate the endonuclease activity residues, which is highly preserved among subtypes of influenza A virus and is able to fold functionally [4]. Translation of viral mRNAs by the host ribosome requires that they are capped at the 5′ end, and this is achieved in cells infected with influenza virus by a “cap-snatching” mechanism, whereby the endonuclease cleaves 5′ caps from host mRNA which then act as primers for transcription. The endonuclease domain binds the N-terminal half of PA (PAN) and contains a two-metal (Mn2+) active site that selectively cleaves the pre-mRNA substrate at the 3′ end of a guanine [3].
The administration of a CAP endonuclease inhibitor, such as Baloxavir marboxil, prevents the above process from occurring, exhibiting its action at the beginning of the pathway before CAP endonuclease may exert its action
It achieves this by inhibiting the process known as cap snatching[4], which is a mechanism exploited by viruses to hijack the host mRNA transcription system to allow synthesis of viral RNAs.
Shionogi, in collaboration with licensee Roche (worldwide except Japan and Taiwan), have developed and launched baloxavir marboxil
In March 2018, Shionogi launched baloxavir marboxil for the treatment of influenza types A and B in Japan . In September 2017, Shionogi was planning to file an NDA in the US; in February 2018, the submission remained in preparation
By September 2016, baloxavir marboxil had been awarded Qualified Infectious Disease Product (QIDP) designation in the US
In March 2017, a multicenter, randomized, double-blind, parallel-group, phase III study (NCT02954354; 1601T0831; CAPSTONE-1) was initiated in the US, Canada and Japan to compare a single dose of baloxavir marboxil versus placebo or oseltamivir bid for 5 days in influenza patients aged from 12 to 64 years of age (n = 1494). The primary endpoint was the time to alleviation of symptoms (TTAS).
PATENTS
JP 5971830
Kawai, Makoto; Tomita, Kenji; Akiyama, Toshiyuki; Okano, Azusa; Miyagawa, Masayoshi
PATENTS
WO 2017104691
Shishido, Takao; Noshi, Takeshi; Yamamoto, Atsuko; Kitano, Mitsutaka
In Japanese Patent Application No. 2015-090909 (Patent No. 5971830, issued on Aug. 17, 2016, Registered Publication), a compound having a CEN inhibitory action and represented by the formula:
[Chemical Formula 2]
is described. Anti-influenza agents of six mechanisms are enumerated as drugs that can be used together with the above compounds. However, no specific combinations are described, nor is it disclosed nor suggested about the combined effect.
Synthesis Example 2
[formula 39]
Compound III-1 (1.00g, 2.07mmol) to a suspension of DMA (5 ml) of chloromethyl methyl carbonate (0.483 g, 3.10 mmol) and potassium carbonate (0 .572 g, 4.14 mmol) and potassium iodide (0.343 g, 2.07 mmol) were added, the temperature was raised to 50 ° C. and the mixture was stirred for 6 hours. Further, DMA (1 ml) was added to the reaction solution, and the mixture was stirred for 6 hours. The reaction solution was cooled to room temperature, DMA (6 ml) was added, and the mixture was stirred at 50 ° C. for 5 minutes and then filtered. 1 mol / L hydrochloric acid water (10 ml) and water (4 ml) were added dropwise to the obtained filtrate under ice cooling, and the mixture was stirred for 1 hour. The precipitated solid was collected by filtration and dried under reduced pressure at 60 ° C. for 3 hours to obtain compound II-4 (1.10 g, 1.93 mmol, yield 93%).
1 H-NMR (DMSO-D 6) δ: 2.91-2.98 (1 H, m), 3.24-3.31 (1 H, m), 3.44 (1 H, t, J = 10.4 Hz) J = 10.8, 2.9 Hz), 4.06 (1 H, d, J = 14.3 Hz), 4.40 (1 H, dd, J = 11.5, 2.8 Hz), 3.73 (3 H, s), 4.00 , 5.67 (1 H, d, J = 6.5 Hz), 5.72 (1 H, d, J = 11.8 Hz), 4.45 (1H, dd, J = 9.9, 2.9 Hz), 5.42 J = 8.0, 1.1 Hz), 7.14 – 7.18 (1 H, m ), 7.23 (1 H, d, J = 7.8 Hz), 7.37 – 7.44 (2 H, m)
PATENTS
JP 6212678
PATENTS
JP 6249434
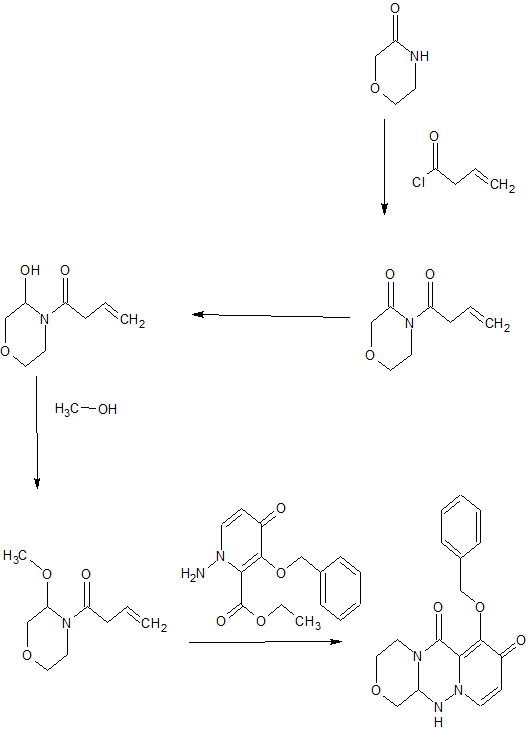
SYNTHESIS OF KEY INTERMEDIATE
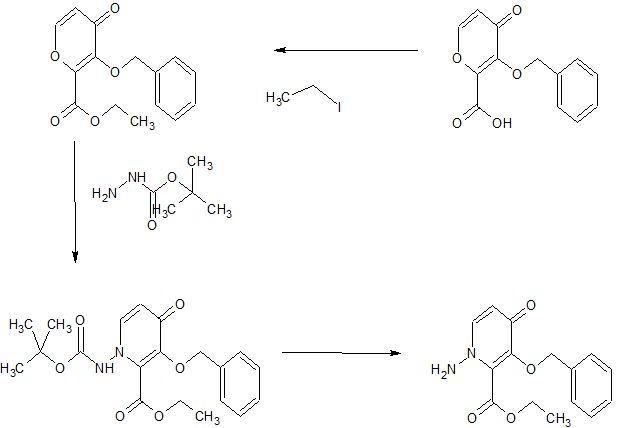
SYNTHESIS OF KEY INTERMEDIATE
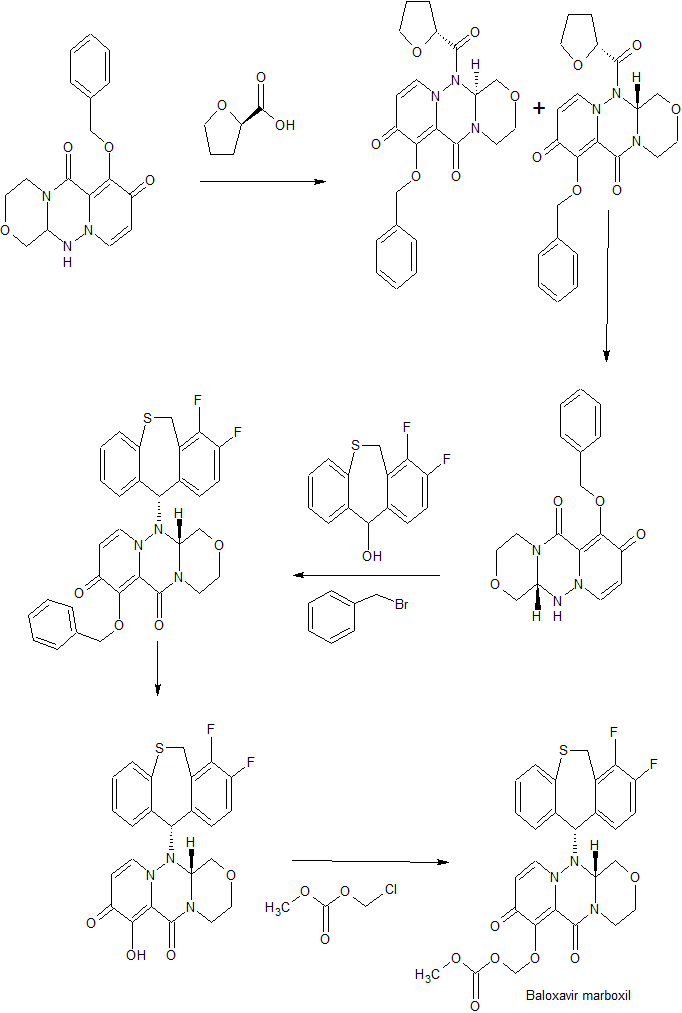
SYNTHESIS OF FINAL PRODUCT
Japan’s New Drug: One Pill May Stop The Flu in Just One Day


Isao Teshirogi, president and chief executive officer of Shionogi & Co., speaks during an interview in Tokyo, Japan. Photographer: Kiyoshi Ota/Bloomberg
One day, you may be able to stop flu viruses in your body in just one day with just one pill. Based on an announcement yesterday, that day may be someday very soon in May in Japan.
On Friday, Japanese pharmaceutical company Shionogi announced that the flu medication that they have developed, Xofluza, otherwise known as baloxavir marboxil (which sounds a bit like a Klingon General), has been approved to be manufactured and sold in Japan. Beginning in October 2015, the medication underwent priority review by Japan’s Ministry of Health, Labor, and Welfare. Shionogi filed for approval in the autumn of 2017. Compared to Tamiflu, which requires two doses each day for five days, apparently only a single dose of Xofluza will be needed to treat the flu. Even though Xofluza has received approval, people will have to wait until the Japanese national insurance sets a price for the medication, which according to Preetika Rana writing for the Wall Street Journal, may not occur until May.
Xofluza works via a different mechanism from neuroaminidase inhibitors like Tamiflu (oseltamivir) and Relenza (zanamivir). Flu viruses are like squatters in your home that then use the furniture and equipment in your home to reproduce. Yes, I know, that makes for a lovely picture. A flu infection begins when flu viruses reach your lungs. Each flu virus will enter a cell in your lungs and then use your cell’s genetic material and protein production machinery to make many, many copies of itself. In order to do this, the flu virus uses “cap-snatching”, which has nothing to do with bottle caps or Snapchat. The virus employs an endonuclease enzyme to clip off and steal the caps or ends of your messenger RNA and then re-purposes these caps to reproduce its own genetic material. After the virus has made multiple copies of itself, the resulting viruses implement another enzyme called a neuroaminidase to separate themselves from parts of the host cell and subsequently spread throughout the rest of your body to cause havoc. While Tamiflu, Relenza, and other neuroaminidase inhibitors try to prevent the neuroaminidase enzyme from working, Xofluza acts at an earlier step, stopping the “cap-snatching” by blocking the endonuclease enzyme.

In a clinical trial, Xofluza stopped an infected person from shedding flu virus sooner than Tamiflu. (Photo Illustration by Ute Grabowsky/Photothek via Getty Images)
By acting at an earlier step before the virus has managed to replicate, Xofluza could stop a flu virus infection sooner than neuroaminidase inhibitors. The results from Shionogi’s Phase III CAPSTONE-1 clinical trial compared Xofluza (then called Cap-dependent Endonuclease Inhibitor S-033188, which doesn’t quite roll off the tongue) with oseltamivir and placebo, with results being published in Open Forum Infectious Diseases. The study found that baloxavir marboxil (or Xofluza) stopped an infected person from shedding flu virus earlier (median 24 hours) than oseltamivir (median 72 hours). Those taking baloxavir marboxil also had lower measured amounts of viruses than those taking oseltamivir throughout the first 3 days of the infection. Baloxavir marboxil also seemed to shorten the duration of flu symptoms (median 53.7 hours compared to a median of 80.2 hours for those taking placebo). Since symptoms are largely your body’s reaction to the flu virus, you can begin shedding virus before you develop symptoms, and symptoms can persist even when you are no longer shedding the virus.
The key with any of these flu medications is early treatment, especially within the first 24 to 48 hours of infection, which may be before you notice any symptoms. Once the virus has replicated and is all over your body, your options are limited. The vaccine still remains the best way to prevent an infection.
In the words of Alphaville, this new drug could be big in Japan. While Xofluza won’t be available in time to help with the current flu season, this year’s particularly harsh flu season has highlighted the need for better ways to treat the flu. But will the United States see Xofluza anytime soon? Similar to Pokemon, Xofluza may need a year or two to reach the U.S. market. But one day, one pill and one day may be a reality in the U.S.
http://www.shionogi.co.jp/en/company/news/2018/pmrltj0000003nx1-att/e180223.pdf
XOFLUZA TM (Baloxavir Marboxil) Tablets 10mg/20mg Approved for the Treatment of Influenza Types A and B in Japan Osaka, Japan, February 23, 2018 – Shionogi & Co., Ltd. (Head Office: Osaka; President & CEO: Isao Teshirogi, Ph.D.; hereafter “Shionogi”) announced that XOFLUZATM (generic name: baloxavir marboxil) tablets 10mg/20mg was approved today by the Ministry of Health, Labour and Welfare for the treatment of Influenza Types A and B. As the cap-dependent endonuclease inhibitor XOFLUZATM suppresses the replication of influenza viruses by a mechanism different from existing anti-flu drugs, XOFLUZATM was designated for Sakigake procedure with priority review by the Ministry of Health, Labour, and Welfare of Japan in October 2015. Shionogi filed for approval to manufacture and sell XOFLUZATM in October 25, 2017. As the treatment with XOFLUZATM requires only a single oral dose regardless of age, it is very convenient, and is expected to improve adherence. XOFLUZATM is expected to be a new treatment option that can improve the quality of life in influenza patients. Shionogi will launch the product immediately after the National Health Insurance (NHI) price listing. Shionogi’s research and development targets infectious disease as one of its priority areas, and Shionogi have positioned “protecting people from the threat of infectious diseases” as one of its social mission targets. Shionogi strives constantly to bring forth innovative drugs for the treatment of infectious diseases, to protect the health of patients we serve.
References
- Jump up^ Rana, Preetika (10 February 2018). “Experimental Drug Promises to Kill the Flu Virus in a Day”. Wall Street Journal.
- Jump up^ “XOFLUZA (Baloxavir Marboxil) Tablets 10mg/20mg Approved For The Treatment Of Influenza Types A And B In Japan”. 23 February 2018 – via www.publicnow.com.
- Jump up^ Dias, Alexandre; Bouvier, Denis; Crépin, Thibaut; McCarthy, Andrew A.; Hart, Darren J.; Baudin, Florence; Cusack, Stephen; Ruigrok, Rob W. H. (2009). “The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit”. Nature. 458(7240): 914–918. doi:10.1038/nature07745. ISSN 0028-0836.
- Jump up^ “Cap snatching”.
 |
|
| Identifiers | |
|---|---|
| CAS Number | |
| PubChem CID | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C27H23F2N3O7S |
| Molar mass | 571.55 g·mol−1 |
| 3D model (JSmol) | |

Shionogi & Company, Limited(塩野義製薬株式会社 Shionogi Seiyaku Kabushiki Kaisha) is a Japanesepharmaceutical company best known for developing Crestor. Medical supply and brand name also uses Shionogi (“シオノギ”).
Shionogi has business roots that date back to 1878, and was incorporated in 1919. Among the medicines produced are for hyperlipidaemia, antibiotics, and cancer medicines.
In Japan it is particularly known as a producer of antimicrobial and antibiotics. Because of antibiotic resistance and slow growth of the antibiotic market, it has teamed up with US based Schering-Plough to become a sole marketing agent for its products in Japan.
Shionogi had supported the initial formation of Ranbaxy Pharmaceuticals, a generic manufacturer based in India. In 2012 the company became a partial owner of ViiV Healthcare, a pharmaceutical company specialising in the development of therapies for HIV.[3]
The company is listed on the Tokyo Stock Exchange and Osaka Securities Exchange and is constituent of the Nikkei 225 stock index.[4]
- Claritin, An anti-histamine marketed in alliance with Schering-Plough.
- Crestor, cholesterol drug
- Nitrazepam, a short-term treatment for insomnia.
- Differin, a topical retinoid for acne.
- Moxifloxacin, antibacterial antiseptic that treats a number of infections
- Cymbalta, an SNRI class anti-depressant, marketed in alliance with Eli Lilly
- Osphena, an estrogen receptor agonist
- Shionogi has a close relationship with Fuji Television Network, Inc., because Shionogi is the sponsor of “Music Fair” (as of 2018, aired on 17 TV stations including TV Oita System Co.) started in 1964.
- Shionogi was a main sponsor of Team Lotus during the age 1991/1994.[5]
- “Shionogi Company Profile”. Retrieved March 18, 2014.
- “Shionogi Annual Report 2013” (PDF). Retrieved March 18, 2014.
- “Shionogi and ViiV Healthcare announce new agreement to commercialise and develop integrase inhibitor portfolio”. viivhealthcare.com. Retrieved 18 March 2014.
- “Components:Nikkei Stock Average”. Nikkei Inc. Retrieved March 11,2014.
- Perry, Alan. “Sponsor Company Profiles”. Retrieved 25 April 2012.
- Official Website (in English)
/////////Baloxavir marboxil, バロキサビルマルボキシル, JAPAN 2018, Xofluza, S-033188, S-033447, RG-6152, Qualified Infectious Disease Product, Priority Review, SAKIGAKE, балоксавир марбоксил , بالوكسافير ماربوكسيل , 玛巴洛沙韦 , Shionogi, roche
COC(=O)OCOC1=C2C(=O)N3CCOCC3N(N2C=CC1=O)C4C5=C(CSC6=CC=CC=C46)C(=C(C=C5)F)F
Polymorphs
https://patents.google.com/patent/WO2017221869A1/en
The Example 3 of Patent Document 1, a manufacturing method of a polycyclic pyridone derivative using pyrone derivatives and pyridone derivative.

The embodiment 165 of Patent Documents 2 and 3, the manufacturing method of the polycyclic pyridone derivative using pyridone derivatives.

However, in the step of coupling an optically active polycyclic pyridone derivatives and thiepine derivatives in Patent Documents 1 to 3, wherein it decrease of optical purity With polycyclic pyridone derivative that is protected with a benzyl group It has not been. On the other hand, no decrease in the optical purity Doing coupling reaction using a polycyclic pyridone derivative protected with a hexyl group, are not described or suggested about that a good yield coupling reaction proceeds. In addition to replace the unsubstituted alkyl group a protecting group from the protected groups other than unsubstituted alkyl group having a polycyclic pyridone derivative reaction, when the reaction is carried out in the presence of magnesium salt, yield and reduction without reaction of the optical purity progress that has not been described or suggested to be.
The process according to Example 21 of Patent Document 1, the step of coupling a polycyclic pyridone derivative and benzhydryl derivatives that are protected with a benzyl group are described. In this manufacturing method, nor is also suggested describe steps of replacing the protecting group of polycyclic pyridone derivative.

The embodiment 175 of Patent Document 2, the step of coupling the tricyclic pyridone derivatives and benzhydryl derivatives substituted are described. In this manufacturing method, not described or suggested for the step of replacing the protecting group of tricyclic pyridone derivatives.

The Examples 583 and 584 of the Patent Document 2, the step of coupling the tricyclic pyridone derivatives and thiepine derivatives substituted are described. In this manufacturing method, not described or suggested about that process and optical purity to replace the protective group of tricyclic pyridone derivatives is reduced.

WO 2010/110409 pamphlet WO 2010/147068 pamphlet WO 2012/039414 pamphlet
Herein, compounds of the formula (V) or (VI) has a cap-dependent endonuclease inhibitory activity, treatment of symptoms and / or disease induced from that infected with influenza virus and / or it is described in PCT / JP2016 / 63139 is useful as a preventive agent.
nternational Publication No. 2012/039414 pamphlet
Example 1 Compound 3

Step 1 Compound 3 of the method for manufacturing compound 1 (100.00g, 406 mmol) was stirred with DMA (300 mL) to. Sodium bicarbonate (44.41 g, 529 mmol), was added dimethyl sulfate (58.91 g, 467 mmol) and DMA (100 mL) was stirred for 7 hours at 25 ° C.. Synthesis hydrochloride to the reaction mixture (16.90 g) and water (500 g) was added, and extracted twice with ethyl acetate (1000,550mL). The organic layer was washed with 5% brine (300 g) and water (300 g). Concentration under reduced pressure, the solvent was distilled off the liquid weight to about 500 g. More ethyl acetate (350 mL) was added to the concentrate, by concentration under reduced pressure, the solvent was distilled off the liquid weight to about 500 g. The DMA (300 mL) was added to the concentrate, and the solvent was distilled off the liquid weight to about 400g by concentration under reduced pressure. After addition of pyridine p-toluenesulfonic acid (265.42g) and DMA (100 mL) to the concentrate, the reaction liquid was heated to 60 ° C.. Was then added slowly over DMA (100 mL) solution of 6 hours carbazate tert- butyl the reaction solution (69.80 g, 528 mmol). The reaction was stirred for 3 hours at 60 ° C., and then cooled to 25 ° C.. After addition of ethanol (100 mL) and water (290 mL) to the reaction mixture and the mixture was heated to 30 ° C.. The reaction solution mixture of ethanol (100 mL) and water (520 mL) was slowly added to. After the reaction solution was cooled to 0 ° C., and stirred for 1.5 hours at 0 ° C.. The resulting pale yellowish white precipitate was collected by filtration. The resulting solid was washed with a mixture of ethanol (480 mL) and water (720 mL), and dried to give monohydrate of Compound 3 (122.70g, 77% yield) as a pale yellowish white solid .
1 H-NMR (400MHz, CDCl 3) δ: 1.45 (s, 9H), 3.77 (s, 3H), 5.26 (s, 2H), 6.39 (d, J = 7.6Hz, 1H), 7.27-7.47 (m , 6H), 7.64-8.23 (br s, 1H)
Powder X-ray diffraction 2θ (°): 5.4,7.5,8.4,10.6,11.9,13.5,20.2,22.9
The powder X-ray diffraction results of Compound 3 shown in FIG.
The water content of the measurement result by Karl Fischer method: 4.5%
Method of Example 2 Compound 9

Step 1 Compound 6 of the method for manufacturing compound 4 (20.00g, 104.6mmol) Compound 5 (28.29g, 167.4mmol) and stirred added DMA (65 mL) to. The reaction solution was heated to 40 ° C., was added sodium tert- butoxide (15.09g, 157.0mmol) slowly. The reaction was stirred for 3 hours at 40 ° C., cooled to 20 ° C.. Acetate (3.14 g) and 10% brine (64 g) was added to the reaction solution, and extracted twice with ethyl acetate (60 mL). The organic layer was added water (144 mL) was cooled to 0 ° C.. The resulting pale yellowish white precipitate was collected by filtration. The resulting solid was washed with a mixture of methanol (5.4 g) and water (48.6 g), and dried to give Compound 6 (20.44 g, 78% yield) as a pale yellowish white solid.
1 H-NMR (CDCl 3) δ: 3.34 (s, 6H), 3.53 (d, J = 5.2Hz, 2H), 3.76 (t, J = 5.6Hz, 2H), 3.90 (t, J = 5.6Hz, 2H), 4.43 (t, J = 5.2Hz, 1H), 7.70-7.73 (m, 2H), 7.84-7.87 (m, 2H)
Production method Compound 6 (20.02 g, 71.68 mmol) of Step 2 compound 8 was stirred with ethanol (20 mL) and water (20 mL) to. The reaction was heated to 60 ° C.. 60% hydrazine monohydrate aqueous solution to the reaction solution (8.99 g, 107.7 mmol) was added, and the mixture was stirred for 4 hours at 60 ° C.. Water (40 mL) was added to the reaction mixture, after cooling to 30 ° C., was added 17% aqueous potassium hydroxide solution (92.12 g). The reaction mixture was extracted 4 times with methylene chloride (120, 78, 78, 78 mL). The organic layer was washed with water (20 mL), and the solvent was distilled off the liquid weight to about 160g by concentration under reduced pressure. In THF (100 mL) was added to the concentrate, and the solvent was distilled off the liquid weight to about 40g by concentration under reduced pressure. In THF (100 mL) was added to the concentrate, and the solvent was distilled off the liquid weight to about 40g by concentration under reduced pressure. In THF (20 mL) was added to the concentrate to give a THF solution of compound 7 15g of a concentrated under reduced pressure.
Compound 3 (10.00g, 25.5mmol) in THF solution of the above compound 7 (14.71 g), were added and stirred THF (7 g) and 1,8-diazabicyclo [5.4.0] -7-undecene (379.0mg) . After the reaction was warmed to 60 ° C., and stirred at 60 ° C. 24 hours. The reaction solution was cooled to 25 ° C., it was added water (28 g) and acetic acid (3.72 g). The reaction solution was extracted twice with ethyl acetate (50,30mL), the organic layer was washed with 5% aqueous sodium hydrogen carbonate solution (30 g) and water (28 g). Reduced pressure and the solvent was distilled off the liquid weight to about 36g Concentration. Ethyl acetate was added to the reaction solution, the solvent was distilled off the liquid weight to about 36g by concentration under reduced pressure. Heptane (65 mL) was added to the concentrate, and cooled to 5 ° C.. 5 After stirring for 1 hour at ° C., was collected by filtration as a pale yellowish white precipitate formed. The resulting solid was washed with a mixture of heptane (32 mL) and ethyl acetate (14 mL), and dried to give Compound 8 (10.10 g, 81% yield) as a pale yellowish white solid.
1 H-NMR (CDCl 3) δ: 1.44 (s, 9H), 3.32-3.48 (m, 12H), 4.41 (t, J = 5.2Hz, 1H), 5.29 (s, 2H), 6.38 (d, J = 7.6Hz, 1H), 7.11-7.50 (m, 7H), 8.46 (s, 1H).
Step 3 Compound 9 of the manufacturing process compound 8 (19.99g, 40.7mmol) in was stirred with acetonitrile (170 mL) and water (30 mL). The reaction mixture was allowed to warm to 60 ° C., methanesulfonic acid (11.70g, 121.7mmol) was added slowly. The reaction mixture was stirred for 6 hours at 60 ° C., and then cooled to 25 ° C.. 30% aqueous solution of sodium hydroxide to the reaction mixture (15.91g) was added, and the solvent was distilled off the liquid weight to about 100g by concentration under reduced pressure. The liquid weight by concentration under reduced pressure by addition of water (50 mL) to the concentrate and the solvent was distilled off to about 100 g. After the concentrated solution was stirred for 30 minutes at 25 ° C., it was collected by filtration a yellow precipitate formed. The resulting solid was washed with water (40 mL), and dried to give 0.5 hydrate of the compound 9 (10.43 g, 76% yield) as yellow crystals.
1 H NMR (400 MHz, DMSO -d 6) δ: 2.95 (ddd, J = 13.7, 12.3, 4.3 Hz, 1H), 3.13 (dd, J = 11.2, 10.0 Hz, 1H), 3.44 (td, J = 11.9, 3.1 Hz, 1H), 3.96-4.08 (m, 2H), 4.14 (dd, J = 13.9, 2.4 Hz, 1H), 4.80 (ddd, J = 12.6, 9.9, 4.5 Hz, 1H), 5.08 (s , 2H), 6.22 (d, J = 7.6 Hz, 1H), 7.24-7.41 (m, 4H), 7.52-7.60 (m, 2H), 7.69 (d, J = 7.6 Hz, 1H)
Powder X-ray diffraction 2θ (°): 9.5,13.4,18.0,19.3,21.2,22.5,22.8,23.6,27.5,28.1
The powder X-ray diffraction results of Compound 9 shown in FIG.
Water content measurement by Karl Fischer: 2.8%
Method of Example 3 Compound 13

0.5 hydrate of the method for manufacturing compound 9 Step 1 Compound 11 and 12 (30.00g, 89.2mmol) in ethyl acetate (87mL), 50 (w / w)% T3P ethyl acetate solution (145.80g, 229.1mmol) It was added and stirred. The reaction mixture was allowed to warm to 60 ° C., after addition of triethylamine (18.55g, 183.3mmol), (R) – (+) – tetrahydrofuran-2-carboxylic acid (12.24g, 105.4mmol) was added slowly. The reaction was stirred for 4 hours at 60 ° C., after cooling to 0 ° C., it was collected by filtration as a pale yellow precipitate formed. By washing the obtained solid with ethyl acetate (120 mL), compound 11 (18.34 g, wet) was obtained as a pale yellow solid. Further, by mixing the filtrate and washings was obtained ethyl acetate solution of compound 12 (358.60g).
Step 2 Compound 13 and 9 of the manufacturing method compound 11 (15.28 g) ethyl acetate (120 mL) to and 1,8-diazabicyclo [5.4.0] -7-undecene (530 mg, 3.5 mmol) was added and stirred. The reaction mixture was heated to 30 ° C., it was slowly added a mixture of methanol (1.67 g) and ethyl acetate (43 mL). The reaction was stirred for 1 hour at room temperature, it was collected by filtration white precipitate formed. The obtained crystals were washed with ethyl acetate (60 mL), and dried to obtain a white crystal of the compound 13 (11.06 g, 45% yield).
1 H-NMR (CDCl3) δ : 2.84-2.92 (m, 2H), 3.45 (td, J = 3.2Hz, 12.0Hz, 1H), 3.82 (dd, J = 4.0Hz, 11.2Hz, 1H), 3.92 ( dd, J = 4.4Hz, 11.6Hz, 1H), 4.13 (dd, J = 2.8Hz, 13.6Hz, 1H), 4.47-4.54 (m, 1H), 4.96 (d, J = 9.6Hz, 1H), 5.27 (d, J = 10.0Hz, 1H), 5.76 (d, J = 13.2Hz, 1H), 6.19 (d, J = 7.6Hz, 1H), 7.22 (d, J = 8.0Hz, 1H), 7.30-7.38 (m, 3H), 7.59 (dd, J = 1.6Hz, 8.0Hz, 2H).
Powder X-ray diffraction 2θ (°): 7.1,14.1,15.1,21.0,21.2,22.9,23.4
The powder X-ray diffraction results of the compound 13 shown in FIG.
Ethyl acetate solution of compound 12 (334.69g) was distilled off under reduced pressure, the solvent was distilled off the liquid weight to about 170 g. The concentrate was stirred and adjusted to 25 ° C.. Acetonitrile (224 mL) to the reaction solution, slowly added water (56 mL) and 24% aqueous sodium hydroxide solution (150 g), was separated into an organic layer and an aqueous layer. To the resulting aqueous layer was added water (14 mL), it was performed twice extracted with acetonitrile (168 mL). The combined organic layers were evaporated under reduced pressure, the solvent was distilled off the liquid weight to about 250 g. The concentrate was heated to 60 ° C., 1,8-diazabicyclo [5.4.0] -7-undecene (19.01g, 124.9mmol) was added. The reaction was stirred for 3 hours at 60 ° C., and then cooled to 40 ° C.. 5.8% aqueous hydrochloric acid (50.40g) was added to the reaction solution to obtain a solution of cooled 314.96 g to 25 ° C.. Some of the solution (158.86g) was concentrated under reduced pressure, the solvent was distilled off the liquid weight to about 85 g. After the concentrated solution was stirred for 2 hours at 20 ° C., was added water (28 mL). The reaction solution was evaporated under reduced pressure concentrated liquid weight to about 100 g. After the concentrated solution was stirred for 1 hour at 20 ° C., the precipitated pale yellowish white crystals were collected by filtration. The obtained crystals were washed with water (42 mL), and dried to give compound 9 (5.93 g, 42% yield) as a pale yellowish white crystals.
Method of Example 4 Compound 19

Step 1 manufacturing method THF (25 mL) of diisopropyl amine compound 15 (7.69 g, 76.0 mmol) added and stirred and cooled to -40 ° C.. The reaction solution 1.6 mol / L n-butyllithium (43.5 ml, 69.6 mmol) After slowly added and stirred for 1 hour at 0 ° C.. Was cooled to -40 ° C., it was slowly added THF (25 mL) solution of 3,4 Jifurooro acid (5.00g, 31.6mmol). The reaction was stirred for 1 hour at -40 ° C., N, it was added N- dimethylformamide (5.74 g, 78.5 mmol) slowly. 6 mol / L aqueous hydrochloric acid (34.25mL) was added to the reaction solution, after heating to 25 ° C., was separated into an organic layer and an aqueous layer. The resulting aqueous layer was extracted with ethyl acetate (15 mL). The organic layers were combined and washed with water (5mL). After concentration under reduced pressure, by adding toluene to the residue, to obtain a toluene solution of Compound 15.
The toluene solution of compound 15 obtained in the manufacturing method of Step 2 compound 16, stirred with toluene (17.8 mL), thiophenol (3.90 g, 35.4 mmol) and D- camphorsulfonic acid (1.16 g, 5.0 mmol) then, the temperature was raised to 60 ℃. The reaction was stirred for 4 hours at 60 ° C., and cooled to 5 ° C.. The reaction solution 2 mol / L aqueous sodium hydroxide solution (10 mL) was added, followed by heating to 25 ° C.. The reaction solution was extracted with toluene (10 mL), the resulting organic layer was washed with 2 mol / L aqueous sodium hydroxide solution (5 mL) and water (10 mL). After concentration under reduced pressure, by addition of toluene to obtain a toluene solution of Compound 16.
Production method of aluminum chloride (5.52 g, 41.4 mmol) of Step 3 compound 17 is stirred with toluene (25 mL) to was cooled to 0 ° C.. To the reaction solution 1,1,3,3 (5.56 g, 41.4 mmol) was added dropwise toluene (10 mL) solution of was heated to 25 ° C.. Toluene solution of compound 16 obtained above to the reaction solution was slowly added, and stirred for 2 hours at 25 ° C.. After 15% aqueous sulfuric acid the (35 mL) was added and stirred the reaction solution was separated into an organic layer and an aqueous layer. The resulting organic layer was washed twice with water (20 mL). Concentration under reduced pressure, the solvent was distilled off the liquid weight to about 16g. Was added slowly heptane (40 mL) to the concentrate, was cooled to 0 ° C., it was collected by filtration white precipitate formed. The resultant solid was washed with heptane (20 mL), and dried to give Compound 17 (7.20 g, 81.3% yield) as a white solid.
1 H-NMR (CDCl 3) δ: 4.61 (d, J = 1.6Hz, 2H), 7.09-7.15 (m, 1H), 7.23-7.27 (m, 3H), 7.34-7.37 (m, 2H), 7.84 -7.88 (m, 1H)
Production method polyphosphoric acid Step 4 Compound 18 (425.0g) was stirred and heated to 80 ° C.. Thereto was added a compound 17 (85.0 g) was heated to 120 ° C., the reaction was stirred for 3 hours at 120 ° C.. The reaction was cooled to 80 ° C., it was slowly added water (200 mL). The reaction solution was cooled to 30 ° C., water (850 mL) was added, and extracted with ethyl acetate (850 mL). The organic layer was washed with water (425 mL) and 10% aqueous sodium bicarbonate solution (255 mL). The solvent was distilled off by a vacuum concentration, it was added heptane (340 mL) to the resulting residue. The solvent was distilled off by a vacuum concentration, it was added heptane (85 mL) to the resulting residue. After stirring for 30 minutes at a reaction solution 30 ° C., it was collected by filtration and the resulting brown precipitate. The resultant solid was washed with heptane (42 mL), and dried to give Compound 18 (72.0 g, 91% yield) as a brown solid.
1 H-NMR (CDCl3) δ : 4.14 (d, J = 1.0Hz, 2H), 7.09-7.18 (m, 1H), 7.27-7.33 (m, 1H), 7.34-7.45 (m, 3H), 8.19 ( dd, J = 8.5Hz, 1.4Hz, 1H)
Step 5 production process of sodium borohydride compound 19 (234.0mg, 6.2mmol) was suspended in a 0.5% aqueous solution of sodium hydroxide (1.8 mL), was prepared sodium borohydride suspension. Compound 18 (4.5g, 17.2mmol) added and stirred in 2-propanol (20 mL) and water (2.25 mL), was heated to 40 ° C.. The was slowly added sodium borohydride suspension prepared in the reaction solution. The reaction was stirred for 1.5 hours at 40 ° C., and cooled to 25 ° C.. Water (32 mL) was added to the reaction solution was further added a mixture of water (6.7 mL) and 62% aqueous sulfuric acid (460 mg). The reaction solution was cooled to 5 ° C., it was collected by filtration and the resulting brown precipitate. The resultant solid was washed with water (18 mL), and dried to give compound 19 (4.4 g, 97% yield) as a brown solid.
1 H-NMR (CDCl3) δ : 2.67 (d, J = 3.8Hz, 1H), 4.20 (dd, J = 14.4, 1.4Hz, 2H), 4.68 (dd, J = 14.5, 1.3Hz, 2H), 7.02 (dt, J = 9.7, 8.3Hz, 1H), 7.12-7.21 (m, 4H), 7.44-7.49 (m, 1H)
Method of Example 5 Compound (V) and Compound (VI)

Production method 1-hexanol (22.5 g, 220 mmol) in Step 1-1 Compound 20 as a mixture of THF (24.6 g), the temperature was adjusted to 20 ° C.. Mixture of isopropyl magnesium chloride THF solution (2mol / L, 7.2g, 14.7mmol) was added to prepare a magnesium hexoxide solution.
Compound 13 (12.0 g, 36.7 mmol) in 1-hexanol (22.5 g, 220 mmol) was stirred with the temperature was adjusted to 20 ° C.. Magnesium prepared above to the resulting slurry hexoxide was added. The reaction was stirred for 4 hours at 20 ° C., and mixed with aqueous citric acid (Citric acid monohydrate 3.1 g, water 36 g). And extracted with THF (10.7 g), the resulting organic layer was washed with water (24 g). Concentration under reduced pressure, the solvent was distilled off the liquid weight to about 55 g. To the resulting solution in THF p-toluenesulfonic acid (p-toluenesulfonic acid monohydrate 7.0 g, tetrahydrofuran 42.8 g) was added. Concentration under reduced pressure, the solvent was distilled off the liquid weight to about 61 g. In THF (42.7 g) was added, by concentration under reduced pressure, the solvent was distilled off the liquid weight to about 61 g. After the temperature was raised to 50 ° C., was added methyl tert- butyl ether (133.0 g), and cooled to 10 ° C.. And stirred 1.5 hours at 10 ° C., it was collected by filtration white precipitate formed. A mixture of methyl tert- butyl ether (40.0 g) and ethyl acetate (16.0 g), washed the resulting solid by drying, the tosylate salt of Compound 20 (15.8 g, 87.2% yield) It was obtained as white crystals.
1 H-NMR (CDCl 3) δ: 0.88 (t, J = 7.2 Hz, 3H), 1.25-1.34 (m, 4H), 1.34-1.43 (m, 2H), 1.76-1.85 (m, 2H), 2.34 (s, 3H), 3.04 (ddd, J = 13.6, 11.7, 4.3 Hz, 3H), 3.36 (dd, J = 11.6, 10.0 Hz, 3H), 3.43 (ddd, J = 13.6, 12.0, 4.4 Hz, 3H ), 4.00 (dd, J = 11.7, 4.3 Hz, 1H), 4.06-4.18 (m, 4H), 4.80 (br, s, 1H), 7.16 (d, J = 7.8 Hz, 1H), 7.62 (d, J = 7.8 Hz, 1H), 7.62 (d, J = 7.1 Hz, 1H), 8.17 (d, J = 7.1 Hz, 1H), 8.40 (br, s, 1H).
Powder X-ray diffraction 2θ (°): 5.9,8.4,11.6,12.7,13.1,15.7
The powder X-ray diffraction results of the compound 20 shown in FIG.
In the manufacturing method above step 1-1 Step 1-2 Compound 20, instead of the THF solution of isopropyl magnesium chloride (0.4 eq), THF solution (16.2wt%, 0.4eq) of cyclohexyl magnesium chloride is reacted with, analysis of the reaction mixture by HPLC, and measured compound 20 production rate.
HPLC area percentage of Compound 20: 90.9% (RT = 11.0min)
Others are the same method as in Step 1-1.
(Measurement condition)
(1) Column: X Select (registered trademark) CSH C18 (3.5μm i.d.4.6x100mm) (Waters)
Flow rate: 1.0 mL / min; UV detection wavelength: 254 nm;
Mobile phase: [A] 0.1% formic acid-containing aqueous solution, subjected to a linear gradient of [B] acetonitrile 5 min, maintaining 15% solvent [B], 15% -60% solvent at 10 min [B], 2 after linear gradient of 60% -85% solvent [B] in minutes, 3 minutes, maintaining 85% solvent [B].
Production method compound 13 (4.91 g, 15.0 mmol) of Step 1-3 Compound 20 in 1-hexanol (27.5 g, 270 mmol) was stirred with the temperature was adjusted to 0 ° C.. The resulting slurry sodium tert- pentoxide in THF (1.4mol / L, 45.0mmol) was added. The reaction was stirred for 2.5 hours at 0 ° C., analysis of the reaction mixture by HPLC, and measured Compound 20 production rate.
HPLC area percentage of Compound 20: 93.3% (RT = 9.5min)
(Measurement condition)
(1) column: CHIRALPAK (registered trademark) IB (5.0μm i.d.4.6x250mm) (DAICEL)
Flow rate: 1.0 mL / min; UV detection wavelength: 254 nm;
Mobile phase: [A] 0.1% formic acid-containing aqueous solution, after a linear gradient of [B] acetonitrile 5 min, maintaining 35% solvent [B], 35% -85% solvent at 6 min [B] , 2 minutes, and maintained 85% solvent [B].
As indicated above, the reaction in good yield when the reaction is carried out using magnesium or sodium salt was found to proceed. It was obtained the desired product in high yield particularly when using isopropylmagnesium chloride.
Step 2 Compound 21 of the method for manufacturing the compounds of the mesylate 20 (12.0g, 24.3mmol) Compound 19 (8.0g, 30.3mmol), was added ethyl acetate (48.7 g) and cyclohexane (14.1 g) stirring at 25 ° C. did. 50 (w / w)% T3P ethyl acetate solution (20.91 g, 32.9 mmol) was added, then methanesulfonic acid (3.5 g, 36.4 mmol) was added. The temperature was raised to 60 ° C., and stirred for 24 hours. After cooling to 25 ° C., it was added THF (32.0 g) and water (24.0 g). Was slowly added 24% aqueous sodium hydroxide solution (30.8 g), it was separated to stand, an organic layer and an aqueous layer. The organic layer was washed twice with 7% saline solution (60.0 g). The resulting solution cyclohexane (9.3 g), ethyl acetate (32.1 g) and methanesulfonic acid (2.80 g, 29.1 mmol) the mixture was added a solution of. Stirred for 2 hours at 25 ° C., it was collected by filtration white precipitate formed. With ethyl acetate (43.3 g), washed the resulting solid, and dried to give the mesylate salt of compound 21 (13.65 g, 84.6% yield) as white crystals.
1 H-NMR (DMSO-d 6) δ: 0.90 (3H, t, J = 6.0 Hz), 1.29-1.36 (4H, m), 1.39-1.49 (2H, m), 1.67-1.79 (2H, m) , 2.38 (3H, s), 2.94 (1H, br s), 3.30 (1H, td, J = 11.6, 2.4 Hz), 3.51 (1H, t, J = 10.4 Hz), 3.66 (1H, dd, J = 11.2, 2.8 Hz), 3.92-4.01 (2H, m), 4.07 (1H, d, J = 14.3 Hz), 4.20 (1H, s), 4.42-4.52 (1H, m), 5.43 (1H, dd, J = 14.4, 2.1 Hz), 5.79-5.83 (2H, m), 6.81 (1H, td, J = 7.6, 1.2 Hz), 6.96 (1H, dd, J = 7.8, 1.0 Hz), 7.09 (1H, J = 8.0, 1.6 Hz), 7.12-7.18 (1H, m), 7.32 (1H, d, J = 7.7 Hz), 7.37-7.49 (2H, m)
Powder X-ray diffraction 2θ (°): 7.1,9.3,12.6,14.1,17.7,18.7,19.2,22.2,25.4,27.7,28 .5,37.8
The powder X-ray diffraction results of the compound 21 shown in FIG.
DSC: Onset 216 ℃, Peak 219 ℃
Production method Compound 21 (15.0g, 22.6mmol) of Step 3 compound (V) were added and stirred to N- methylpyrrolidone (52.4 g). Lithium chloride (8.6g, 203.3mmol) was heated to addition 75 ° C. to. Was stirred at 75 ° C. 20 h, then cooled to 40 ° C.. Acetonitrile (20.0 g) was added, was further added water (11.6 g). Cooled to 30 ° C., followed by stirring for 30 minutes, water was slowly added (142.5 g). After stirring 1.5 hours at 30 ° C., it was collected by filtration white precipitate formed. The resulting solid was washed with 2-propanol (60.1 g), and dried to give compound (V) (9.91g, 90.7% yield) as white crystals.
1 H-NMR (CDCl3) δ : 3.00 (td, J = 11.8, 3.2 Hz, 1H), 3.46 (td, J = 12.0, 2.8 Hz, 1H), 3.59 (t, J = 10.0 Hz, 1H), 3.82 (dd, J = 12.2, 3.0 Hz, 1H), 3.96 (dd, J = 11.0, 3.0 Hz, 1H), 4.07 (d, J = 13.6 Hz, 1H), 4.58 (dd, J = 10.0, 2.8 Hz, 1H), 4.67 (dd, J = 13.6, 2.0 Hz, 1H), 5.26-5.30 (m, 2H), 5.75 (d, J = 8.0 Hz, 1H), 6.69 (d, J = 7.6 Hz, 1H), 6.83-6.87 (m, 1H), 6.99-7.04 (m, 2H), 7.07-7.15 (m, 3H).
Powder X-ray diffraction 2θ (°): 9.6,10.9,17.8,21.5,22.1,23.5,24.8
Compound powder X-ray diffraction results of (V) shown in FIG.
Step 4 Compound preparation method a compound of (VI) (V) (1.00g, 2.07mmol) to a suspension of DMA (5 ml) of chloromethyl methyl carbonate (0.483 g, 3.10 mmol) and potassium carbonate (0.572 g, 4.14 mmol), potassium iodide (0.343 g, 2.07 mmol) was added and stirred heated 6 hours to 50 ° C.. Furthermore, the liquid reaction mixture was stirred for 6 hours added DMA (1 ml). The reaction mixture was cooled to room temperature, and stirred for 5 minutes at 50 ° C. was added a DMA (6 ml), and filtered. The obtained filtrate under ice-cooling, was added dropwise 1 mol / L aqueous hydrochloric acid (10ml) and water (4 ml), and stirred for 1 hour. The precipitated solid was collected by filtration, subjected to 3 hours under reduced pressure dried at 60 ° C., to obtain a compound (VI) (1.10g, 1.93mmol, 93% yield).
1H-NMR (DMSO-D6) δ: 2.91-2.98 (1H, m), 3.24-3.31 (1H, m), 3.44 (1H, t, J = 10.4 Hz), 3.69 (1H, dd, J = 11.5, 2.8 Hz), 3.73 (3H, s), 4.00 (1H, dd, J = 10.8, 2.9 Hz), 4.06 (1H, d, J = 14.3 Hz), 4.40 (1H , d, J = 11.8 Hz), 4.45 (1H, dd, J = 9.9, 2.9 Hz), 5.42 (1H, dd, J = 14.4, 1.8 Hz), 5.67 (1H, d, J = 6.5 Hz), 5.72 -5.75 (3H, m), 6.83-6.87 (1H, m), 7.01 (1H, d, J = 6.9 Hz), 7.09 (1H, dd, J = 8.0, 1.1 Hz), 7.14-7.18 (1H, m ), 7.23 (1H, d, J = 7.8 Hz), 7.37-7.44 (2H, m).
1 H-NMR (DMSO-D 6) δ: 2.91-2.98 (1H, m), 3.24-3.31 (1H, m), 3.44 (1H, t, J = 10.4 Hz), 3.69 (1H, dd, J = 11.5, 2.8 Hz), 3.73 (3H, s), 4.00 (1H, dd, J = 10.8, 2.9 Hz), 4.06 (1H, d, J = 14.3 Hz), 4.40 (1H, d, J = 11.8 Hz) , 4.45 (1H, dd, J = 9.9, 2.9 Hz), 5.42 (1H, dd, J = 14.4, 1.8 Hz), 5.67 (1H, d, J = 6.5 Hz), 5.72-5.75 (3H, m), 6.83-6.87 (1H, m), 7.01 (1H, d, J = 6.9 Hz), 7.09 (1H, dd, J = 8.0, 1.1 Hz), 7.14-7.18 (1H, m), 7.23 (1H, d, J = 7.8 Hz), 7.37-7.44 (2H, m).