















BMS-599793
(DS003, BMS-599793)
Cas 676489-50-2
| Molecular Formula: | C27H22N6O3 |
|---|---|
| Molecular Weight: | 478.512 g/mol |
2-[1-[2-(4-methoxy-7-pyrazin-2-yl-1H-pyrrolo[2,3-c]pyridin-3-yl)-2-oxo-acetyl]-4-piperidylidene]-2-phenyl-acetonitrile;
2-[1-[2-(4-methoxy-7-pyrazin-2-yl-1H-pyrrolo[2,3-c]pyridin-3-yl)-2-oxoacetyl]piperidin-4-ylidene]-2-phenylacetonitrile;
Piperidine, 4-(cyanophenylmethylene)-1-[2-(4-methoxy-7-pyrazinyl-1H-pyrrolo[2,3-c]pyridin-3-yl)-1,2-dioxoethyl]-;
US 20040063744 , US 20040186292, WO 2009114313
![]()
| Inventors | Andrew S. Thompson, Hua Cheng, Stanislaw Pikul |
| Applicant | International Partnership For Microbicides |

2-(1-(2-(4-methoxy-7-(pyrazin-2-yl)-1H-pyrrolo[2,3-c]pyridin-3-yl)-2-oxoethanoyl)piperidin-4- ylidene)-2-phenylethanenitrile
1 (DS003, BMS-599793) is a small molecule entry inhibitor that interferes with HIV infection by binding to the gp120 protein.1 The International Partnership for Microbicides (IPM) licensed 1 from Bristol-Myers Squibb (BMS) with the goal to develop it as a topical microbicide for use in resource-poor countries. Microbicides are vaginal dosage forms of potent inhibitors of HIV that women can use to prevent sexual transmission of HIV from male partners.
1 (a) Maddon, P. J.; Dalgleish, A. G.; McDougal, J. S.; Clapham, P. R.; Weiss, R. A.; Axel. R. Cell 1986, 47, 333-348. (b) McDougal, J. S.; Kennedy, M. S.; Sligh, J. M.; Cort, S. P.; Mawle, A.; Nicholson, J. K. Science 1986, 231, 382-385. (c) Moore, J. P.; Jameson, B. A.; Weiss, R. A.; Sattentau, Q. J. in Viral fusion mechanisms. ed. J. Bentz, CRC Press, Boca Raton, Fla. 1993, p. 233-289.

The discovery and development of new therapeutic strategies against HIV has extended and improved the quality of life of infected patients. Thus far, 30 antiretroviral drugs have been approved by the Food and Drug Administration to treat individuals infected with HFV. These drugs fall into three major classes: reverse transcriptase inhibitors, protease inhibitors, and entry inhibitors, including fusion inhibitors. Unfortunately, currently available therapies have several limitations.
For example, as HIV reproduces itself, different strains of the virus emerge, some of which are resistant to antiretroviral drugs. Therefore, doctors recommend patients infected with HIV take a combination of antiretroviral drugs known as highly active antiretroviral therapy (HAART). This strategy, which typically combines at least three effective antiretroviral drugs from at least two different classes, has been shown to effectively suppress the virus when used properly.
Patients taking antiretroviral drugs, however, often have low adherence to complicated drug regimens. The currently recommended HAART regimen involves taking several antiretroviral drugs each day, some of which may require fasting and cause unpleasant side effects such as nausea and vomiting. In addition, antiretroviral drugs may cause more serious medical problems, including metabolic changes such as abnormal fat distribution, abnormal lipid and glucose metabolism, and bone loss. Additional problems associated with current therapies include drug-drug interactions, toxicity, poor tolerability, inconvenient dosing frequency, and food interactions, Thus, simpler, less toxic, and more effective drag regimens would be beneficial.
Entry inhibitors represent the newest generation of antivirals for the treatment of HIV. These inhibitors may prove beneficial for the growing number of HIV-infected individuals who have developed resistance to the currently available reverse transcriptase inhibitors and protease inhibitors. These compounds act by interfering with attachment of HIV gpl20 to either the CD4 T cell receptor or the CCR5/CXCR4, thereby blocking entry of the vims into the host cell (Biia «t al, J, Antinύcrβb, Chemother. 57(4):619 (2006)). Maraviroc and enfuvirtide are currently the only entry inhibitors that have been approved by the Food and Drug Administration (FDA). Thus, new entry inhibitors and efficient and effective methods for synthesizing them are needed in the art.



2-(1-(2-(4-methoxy-7-(pyrazin-2-yl)-1H-pyrrolo[2,3-c]pyridin-3-yl)-2-oxoethanoyl)piperidin-4- ylidene)-2-phenylethanenitrile (1, laboratory scale process). A flask was charged with acid 11 (9.29 g, 31.2 mmol), DIPEA (12.9 mL, 78 mmol), 4 (7.18 g, 36.3 mmol) and DMF (95 mL) subsequently. HATU (13.66 g, 35.9 mmol) was added to the reaction mixture over 10 minutes, which was accompanied by increase of internal temperature from 19 0C to 27 0C. After the reaction mixture was stirred at 25 0C for 3.5 h the HPLC analysis showed complete disappearance of acid 11. Ethanol (950 mL) was added and the resulting suspension was heated at reflux for 1 h. The mixture was then cooled to 25 0C and 1 was isolated by filtration and washed with ethanol (50 mL). The material was dried under vaccum at 50 0C to afford 10.58 g (71% yield) of 1 as a colorless solid.
1H NMR (300 MHz, CDCl3) 2.58-2.65 (m, 2H), 2.91-2.99 (m, 2H), 3.48-3.51(m, 1H), 3.68-3.78 (m, 2H), 3.95-3.99 (m, 1H), 4.11 (s, 3H), 7.27-7.46 (m, 5H), 8.16 (d, J = 5.1 Hz, 1H), 8.21-8.25 (m, 1H), 8.60 (s, 2H), 9.82 (d, J = 3.9 Hz, 1H), 11.75(br s, 1H).
LCMS: m/e 479.3 (M+H)+. Analysis by ICP-MS showed 16 ppm Pd, 79 ppm Fe, 102 ppm Zn. This material was found to be a mixture of two polymorphs: Form 1 and Form 2.
kilo-lab scale process including polymorph conversion
1 (84% yield) and 99.6% purity by HPLC. This material was a pure Form 1 polymorph.
1H NMR (400 MHz, CDCl3) 2.61 (t, J = 5.8 Hz, 1H), 2.65 (t, J = 5.9 Hz, 1H), 2.94 (t, J = 5.8 Hz, 1H), 2.99 (t, J = 5.9 Hz, 1H), 3.51 (t, J = 5.8 Hz, 1H), 3.72 (t, J = 5.8 Hz, 1H), 3.78 (t, J = 6.0 Hz, 1H), 3.98 (t, J = 6.0 Hz, 1H), 4.12, 4.12 (two s, 3H), 7.29-7.47 (m, 5H), 8.16, 8.18 (two s, 1H), 8.23, 8.25 (two d, J = 3.1 Hz, 1H), 8.60-8.63 (m, 2H), 9.83, 9.84 (two d, J = 1.4 Hz, 1H), 11.76, 11.78 (two br s, 1H);
13H NMR (100 MHz, CDCl3) 30.3, 31.0, 33.7, 34.3, 41.7, 42.0, 46.0, 46.3, 56.8, 111.4, 115.0 (2C), 117.7 (2C), 120.9, 124.2, 128.9 (2C), 129.0, 129.1 (2C), 131.9, 132.6 (2C), 133.9 (2C), 136.6, 142.2, 143.4, 143.7 (2C), 151.1, 151.3, 154.8, 166.4, 166.5, 185.6
Anal. Calcd for C27H22N6O3: C, 67.77; H, 4.63; N, 17.56. Found: C, 67.84; H, 4.64; N, 17.56.
1H NMRPREDICT


13C NMR PREDICT


Patent
https://www.google.com/patents/WO2009114313A2?cl=en
EXAMPLES
10 Example 1: Synthesis of Iodopyrazine (1) from Chloropyrazine
NaI, HOAc, H2SO4, MeCN, f N reflux,4-6h, ca. 58% f Υ
A reaction mixture of chloropyrazine (7.5 ml, 83 mmol), NaI (30.3 g, 202 15 mmol), HOAc (9.6 ml, 168 mmol) and H2SO4(0.5 ml) in MeCN (105 ml) was heated at reflux for 4.5 hours. The solvent was removed and water (120 ml) was added. After the solution was basified with saturated NaHCO3, it was extracted with dichloromethane (DCM) (2 x 125 ml). The DCM layers were combined, washed with saturated Na2S2O3, brine and dried. The removal of solvent gave crude iodopyrazine as an oil (12.33 g, 20 71%). Analysis by 1H NMR showed there was less than about 10 mol% of chloropyrazine in the oil. Another batch of chloropyrazine (50 g, 437 mmol) was also converted into crude iodopyrazine (about 65 g) by the same procedure. These two batches of crude iodopyrazine were combined and distillation of the crude iodopyrazine under reduced pressure (about 0.75 torr, bp 47°C) gave pure compound 64 g (60%). 25
1H-NMR (CDCl3, 300MHz) 8.40 (dd, /=1.8, 2.4Hz, IH), 8.51 (d, /=2.4Hz,lH), 8.87 (d, /=1.5Hz,lH).
Example 2: Synthesis of Coupled Azaindole (3) from Iodopyrazine (1)
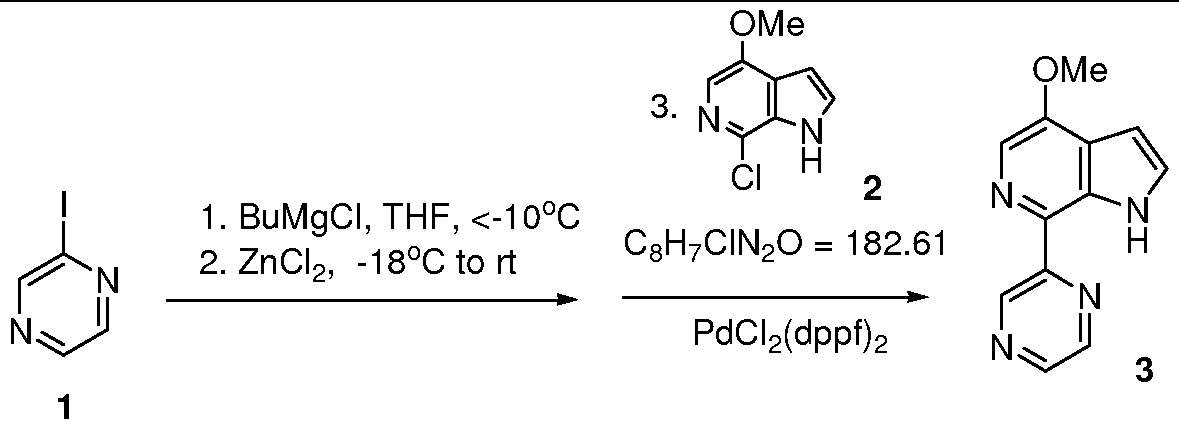
O Q C4H3IN2 = 205.98 C12H10N4O = 226.23 To a solution of iodopyrazine 1 (45.8 g, 0.222 mol) in tetrahydrofuran (THF) (460 ml) at -18°C, BuMgCl (2 M in THF, 108 ml, 216 mmol) was added dropwise via an addition funnel over 20 minutes. The internal temperature of the resulting suspension was raised to -1O0C after addition. The mixture was stirred for another 40 minutes during which time the internal temperature dropped to -180C. Then, ZnCl2 (0.5 M in THF, 220 mmol) was added via addition funnel over 15 minutes. The NaCl-ice bath was removed after addition and the mixture was warmed up to room temperature over 2 hours and was stirred at room temperature for another 0.5 hours. Chloroazaindole 2 (12.95 g, 71 mmol) and PdCl2(dppf)2 (5.8 g, 7.1 mmol) were added into the mixture and mixture heated at 58°C for 6 hours, then stirred at room temperature overnight. Analysis by HPLC showed >20:l ratio of product to starting material.
The reaction was quenched with NH4Cl (36 N aqueous, 25 ml) and the resulting inorganic salt was filtered off and washed with THF. The filtrate was concentrated to about 200 ml and IL of dichloromethane was added. The solution was washed with brine (3×500 ml) and dried (Na2SO4). The solution was concentrated and the residue was absorbed onto silica gel (25 g), then put on top of a silica gel (105 g) column and eluted with hexanes and EtOAc(hexanes:EtOAc=3:l to 0:1). Removal of the solvent gave crude coupled azaindole 3 which was then heated in refluxing EtOAc (350 ml) for about 0.5 hours. After an insoluble sparkling dark red solid was filtered off, and EtOAc was removed, a brown solid (14.86 g) was obtained, which was then dissolved in a refluxing solution of hexanes (40 ml) and EtOAc (120 ml). The resulting solution was cooled to room temperature and the product isolated by filtration to give a brown solid 3 (9.56 g, >99% pure by HPLC, 60% yield).
1H NMR (DMSO-d6, 300 MHz) (δ, ppm): 4.02(s, 3H), 6.63-6.65(m, IH), 7.56(t, / = 2.7Hz, IH), 8.04(s, IH), 8.64(d, / = 2.7Hz, IH), 8.74-8.75(m, IH), 9.62(d, / = 1.5Hz, IH), 11.78 (br, s, IH); LCMS: m/e 227 (M+H)+.
Analysis by ICP-MS showed <1 ppm tin, 1669 ppm iron, 83ppm zinc.
Example 3: Synthesis of Acylated Azaindole (4) from Coupled Azaindole (3)

C12H10N4O = 226.23 C15H12N4O4 = 312.28
3 4
To a solution of dichloromethane and nitromethane (4:1, 200 ml) in a 500 ml 3- neck flask cooled with ice-water bath, was added AlCl3 (22.3g, 168 mmoles) in portions. Then, 3 (4.75 g, 21.0 mmol) was added into the solution in portions. The internal temperature was raised from 1°C to 60C then back tol°C. ClCOCO2Me (3.9ml, 41.1 mmoles) was added into the solution dropwise using a syringe in over about 5 minutes. The resulting homogeneous solution was stirred at 00C for 10 minutes and then put in the cold room (about 00C ) for 15 hours without stirring. Analysis by HPLC after 15 hours showed that the ratio of 3:4:5 was 0:92:3. The reaction solution was then poured into cold 25% aqueous NH4OAc solution (500 ml) in portions. The organic layer was separated and the aqueous layer was extracted with DCM (300 ml, then 2×150 ml). The combined organic layers were washed with brine (2×300 ml) and dried (Na2SO4). Removal of solvent in vacuo gave ester 4 as a solid (4.85 g, 74%).
Analysis by ICP-MS showed <1 ppm tin, 1535 ppm iron, 103ppm zinc.
Example 4: Synthesis of Acylated Azaindole (5) from Acylated Azaindole (4)

C15H12N4O4 = 312.28 C14H10N4O4 = 298.25 4 5
To suspension of ester 4 (10.00 g, 32.1 mmol) in methanol (150 ml), K2CO3 (1
M, 150 ml, 150 mmol) was added. After the reaction mixture was stirred at room temperature for 1 hour methanol was removed in vacuo. The remaining reaction mixture was diluted with water to 1.2 L and washed with MTBE (2×400 ml). The aqueous phase was acidified with HCl (2 M, 185 ml, 370 mmol) to pH=l. The acid 5 (a grey solid) thus formed was filtered off and dried (9.29 g, 97% yield).
Analysis by ICP-MS showed <1 ppm tin, 143 ppm Fe, 96 ppm Zn.
Example 5: Synthesis of Nitrile 6 from l-Boc-4-piperidone
1 ) NaHMDS, THF,
2) TFA, 3)NaHCO3,
Boc-N >=O 4) HCI . HHCCII


6
NaHMDS (2 M in THF, 8.6 ml, 17.2 mmol) was added into a solution of 1-Boc- 4-piperidone (3.0 g, 14.4 mmol) and benzyl cyanide (2.0 ml, 17.2 mmol) in THF (60 ml) at room temperature. The reaction mixture was heated at 50-600C (oil bath) until benzyl cyanide was consumed (as monitored by HPLC). The reaction was quenched by the addition of methanol (12 ml), and the solvent was removed in vacuo. TFA (30 ml, 402 mmol) was added to the residue and the resulting mixture was stirred at room temperature overnight. Most of the TFA was removed in vacuo and saturated NaHCO3 (100 ml) was added. The mixture was extracted with EtOAc (80 ml, 3×30 ml). The organic layers were combined and washed with brine, and dried (Na2SO4). After removal of the EtOAc, the remaining residue was dissolved in DCM (20 ml). The DCM solution was added dropwise to HCl (0.5 M in ether, 40 ml of 2M diluted to 160 ml with ether) at room temperature to form the hydrochloride salt of nitrile 6. The hydrochloride salt of nitrile 6 was then filtered off and was washed with ether (3×10 ml), and dried to afford 2.75 g of a yellow solid (81% yield).
xample 6: Synthesis of DS003 from Acylated Azaindole 5 and Nitrile 6

5 DS003
A 2 L flask was charged with acid 5 (9.29 g, 31.2 mmol), DIPEA (12.9 ml, 78 mmol), nitrile 6 (7.18 g, 36.3 mmol) and DMF (95 ml) subsequently. HATU (13.66 g, 35.9 mmol) was added the reaction mixture in portions over 10 minutes. The internal temperature rose to 27°C from 19°C. After the reaction mixture was stirred at room temperature for 3.5 hours, analysis by HPLC showed that the starting material was completely consumed. Ethanol (950 ml) was added and the resulting suspension was heated at reflux for lhour. The mixture was then cooled to room temperature and
DS003 was isolated by filtration and washed with ethanol (50 ml). The material was dried on a rotovap at 40-500C then by using an oil pump at room temperature to afford 10.58 g of DS003 (71% yield, >99% purity by HPLC).
1H NMR (CDCl3, 300 MHz) (δ, ppm): 2.58-2.65 (m, 2H), 2.91-2.99 (m, 2H), 3.48- 3.51(m, IH), 3.68-3.78 (m, 2H), 3.95-3.99 (m, IH), 4.11 (s, 3H), 7.27-7.46 (m, 5H), 8.16 (d, J = 5.1Hz, IH), 8.21-8.25 (m, IH), 8.60 (s, 2H), 9.82 (d, J = 3.9 Hz, IH), 11.75 (br, IH); LCMS: m/e 479.3 (M+H)+.
Analysis by ICP-MS showed <1 ppm tin, 16 ppm Pd, 79 ppm iron, 102 ppm zinc.
Paper
Synthetic Process Development of BMS-599793 Including Azaindole Negishi Coupling on Kilogram Scale


Abstract

A new approach to the synthesis of 1 (DS003, BMS-599793), a small-molecule HIV entry inhibitor, is described. The initial medical chemistry route has been modified by rearranging the sequence of synthetic steps followed by replacement of the Suzuki coupling step by the Negishi conditions. Acylation of the resulting azaindole 7 under the Friedel–Crafts conditions is studied using monoesters of chlorooxalic acid in the presence of aluminum chloride. Polymorphism of 1is also investigated to develop conditions suitable for preparation of the desired Form 1 of the target compound. The new route is further optimized and scaled up to establish a new process that is applied to the synthesis of kilogram quantites of the target active pharmaceutical ingredient.
| Patent ID | Date | Patent Title |
|---|---|---|
| US7915283 | 2011-03-29 | INDOLE, AZAINDOLE AND RELATED HETEROCYCLIC 4-ALKENYL PIPERIDINE AMIDES |
| US7348337 | 2008-03-25 | Indole, azaindole and related heterocyclic 4-alkenyl piperidine amides |
////////BMS-599793, BMS 599793, DS003, 676489-50-2
O=C(C(=O)c3cnc2c3c(OC)cnc2c1cnccn1)N4CC/C(CC4)=C(\C#N)c5ccccc5
or
COC1=CN=C(C2=C1C(=CN2)C(=O)C(=O)N3CCC(=C(C#N)C4=CC=CC=C4)CC3)C5=NC=CN=C5