















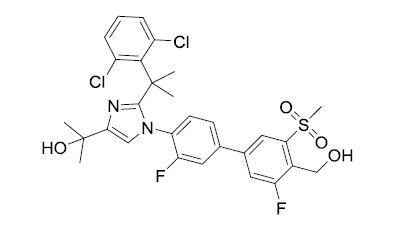
BMS-852927
CAS 256918-39-4
609.51 MW
C29 H28 Cl2 F2 N2 O4 S MF
2-(2-(2-(2,6-dichlorophenyl)propan-2-yl)-1-(3,3′-difluoro-4′-(hydroxymethyl)-5′-(methylsulfonyl)biphenyl-4-yl)-1H-imidazol-4-yl)propan-2-ol
1H-Imidazole-4-methanol, 2-[1-(2,6-dichlorophenyl)-1-methylethyl]-1-[3,3′-difluoro-4′-(hydroxymethyl)-5′-(methylsulfonyl)[1,1′-biphenyl]-4-yl]-α,α-dimethyl-
Treat metabolic syndrome

Brett Busch, Ph.D.
https://www.linkedin.com/in/brettbbusch

| Brett B. Busch, William C. Stevens, Jr., Ellen K. Kick, Haiying Zhang, Venkataiah Bollu,Richard Martin, Raju Mohan | |
| Applicant | Exelixis, Inc. |
| Brett B. Busch, William C. Stevens, JR., Ellen K. Kick, Haiying Zhang, Venkataiah Bollu,Richard Martin, Raju Mohan | |
| Bristol-Myers Squibb Company, Exelixis Patent Company Llc |
- Originator Exelixis
- Developer Bristol-Myers Squibb
- Class Antihyperlipidaemics; Small molecules
- Mechanism of Action Liver X receptor modulators
- Discontinued Atherosclerosis; Hypercholesterolaemia
Most Recent Events
- 04 Jun 2014 BMS 852927 is still in phase I trials for atherosclerosis and in preclinical development for hypecholesterolaemia in USA
- 02 Aug 2013 Bristol-Myers Squibb terminates the planned phase I trial for Hypercholesterolaemia in Germany, Canada and Switzerland (NCT01651273)
- 06 Jul 2012 Bristol-Myers Squibb plans a phase I trial for Hypercholesterolaemia in Germany, Canada and Switzerland (NCT01651273)
1H-NMR (DMSO-d6, 400 MHz) δ 7.94 (m, 2H), 7.63 (dd, 1H, J = 11.29, 1.51 Hz), 7.34 (d, 1H, J = 9.54
Hz), 7.14 (m, 3H), 7.05 (m, 1H), 6.83 (s, 1H), 5.58 (t, 1H, J = 5.27 Hz), 4.96 (d, 2H, J = 4.27 Hz), 4.70
(s, 1H), 3.46 (s, 3H), 1.96 (s, 6H), 1.45 (s, 6H); MS m/e 609.16 (M+H+);
13CNMR (DMSO-d6, 400MHz) 161.42 (d, J=249.49 Hz), 156.85 (d, J=250.25 Hz), 153.18, 148.39, 141.69 (d, J=3.05 Hz), 139.45 (dd, J=9.16, 1.53 Hz), 139.32 (dd, J=8.39, 1.53 Hz), 138.58, 134.68, 131.39, 129.96, 128.40,
127.12 (d, J=17.55 Hz), 125.72 (d, J=12.97 Hz), 123.15 (d, J=2.29 Hz), 122.49 (d, J=3.05 Hz), 119.04
(d, J=25.18 Hz), 116.30, 114.52 (d, J=22.13 Hz), 68.11, 51.97 (d, J=5.34 Hz ), 45.53, 44.78, 44.29,
31.01, 30.53.
19F-NMR (JEOL 500 MHz, CDCl3) -113.55, -116.73.
HPLC (XBridge 5μ C18 4.6x50mm, 4 mL/min, Solvent A: 10 % MeOH/water with 0.2 % H3PO4, Solvent B: 90 % MeOH/water with0.2 % H3PO4, gradient with 0-100 % B over 4 minutes): 2.56 minutes, Purity, 99.7%.
HRMS (m/z,Obs.): 609.12065 [M+H]+; (Calc.): 609.11877. Formula: C29H29Cl2F2N2O4S. Anal. Calcd. for
C29H28N2O4SCl2F2•0.10 C2H6O•0.10 C4H5O2: C, 57.05; H, 4.75; Cl, 11.42; F, 6.10; N, 4.50; S, 5.15.
Found: C, 57.14; H, 4.54; Cl, 11.57; F, 5.94; N, 4.36; S, 5.07. The residual solvents, ethyl acetate (1.39
weight %), ethanol (0.74 weight %), dichloromethane (0.05 weight %), and heptane (< 0.05 weight %)
were identified in the sample by GC/MS and the retention times were matched with the reference standards.


Liver X receptors (LXRs) belong to a family of nuclear hormone receptors that are endogenously activated by cholesterol and its oxidized derivatives to mediate transcription of genes involved in maintaining glucose, cholesterol, and fatty acid metabolism. LXRa is found predominantly in the liver, with low levels found in kidney, intestine, spleen, and adrenal tissue. LXRp is ubiquitous in mammals and was found in nearly all tissues examined. Given the intricate link between lipid metabolism and cancer cell growth, the ubiquitous expression of LXRp in some types of cancer is unlikely to be coincidental, allowing cancer cells to synthesize lipids and lipoprotein particles to sustain their growth. At the same time, however, such stable basal expression levels make LXRp an ideal therapeutic target.

Examples of LXR agonists reported in the literature
PATENT
WO 2010138598
PATENT
WO 2012135082
PATENT
WO 2014028461
PATENT
WO 2016100619
PATENT
https://www.google.com/patents/US8618154?cl=enIt

Example 9 2-(2-(2-(2,6-dichlorophenyl)propan-2-yl)-1-(3,3′-difluoro-4′-(hydroxymethyl)-5′-(methylsulfonyl)biphenyl-4-yl)-1H-imidazol-4-yl)propan-2-ol

Example 9a Preparation of 2-(2,6-dichlorophenyl)-2-methylpropanenitrile
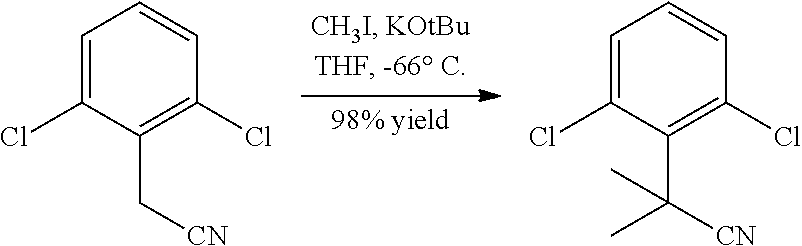
To a 1 M solution of potassium tert-butoxide (403 mL, 403 mmol) at −66° C. (acetone/dry ice) was slowly added 2-(2,6-dichlorophenyl)acetonitrile (25.0 g, 134 mmol) in anhydrous THF (150 mL). The mixture was stirred at −66° C. for 20 minutes. Then, iodomethane (33.6 mL, 538 mmol) was added drop-wise over 25 minutes at −66° C. At this stage, it was exothermic and a large amount of light yellow precipitate was observed. The suspension was stirred at −60° C. for 30 minutes. The reaction mixture was quenched with 200 mL ice water, and extracted with ether (3×150 mL). The organics were combined, washed with 150 mL brine, dried over Na2SO4, and concentrated on a rotary evaporator. The crude product (30 g, yellow oil) was purified by column chromatography (ISCO, 330 g silica, 20% EtOAc in hexanes) to afford 2-(2,6-dichlorophenyl)-2-methylpropanenitrile (28.2 g, 132 mmol, 98% yield) as a light yellowish oil. 1H-NMR (CDCl3, 400 MHz) δ 7.35 (d, 2H, J=8.03 Hz), 7.16 (t, 1H, J=8.0 Hz), 2.09 (s, 6H); 13C-NMR (CDCl3, 126 MHz) δ134.6, 133.8, 131.4, 129.0, 124.1, 38.6, 29.2; MS m/e 214.10 (M+H+); HPLC (XBridge 5μ C18 4.6×50 mm, 4 mL/min, Solvent A: 10% MeOH/water with 0.2% H3PO4, Solvent B: 90% MeOH/water with 0.2% H3PO4, gradient with 0-100% B over 4 minutes): 3.16 minutes.
Example 9b Preparation of N-(4-bromo-2-fluorophenyl)-2-(2,6-dichlorophenyl)-2-methylpropanimidamide

2-(2,6-Dichlorophenyl)-2-methylpropanenitrile (20 g, 93 mmol) and 4-bromo-2-fluoroaniline (28.4 g, 149 mmol) were dissolved in anhydrous o-xylene (200 mL) and heated to 100° C. under N2. Trimethylaluminum (2 M) in toluene (140 mL, 280 mmol) was added drop-wise (˜0.9 mL per minute) over 2.5 hours while the reaction mixture was stirred at 100° C. After addition, the reaction mixture was stirred at 100° C. for 30 minutes, and then cooled to −5° C. The reaction mixture was very carefully quenched with potassium sodium tartrate (20 g in 100 mL water) (Caution: gas and heat formation). The reaction mixture was filtered through Celite 545. The filtrate was washed with 1N HCl (4×70 mL). The aqueous was neutralized with 2N NaOH and extracted with EtOAc (4×100 mL). The organics were combined, washed with brine, dried with Na2SO4, and concentrated on a rotary evaporator to afford 24 g of crude product. The crude product was recrystallized with 72 mL of MTBE and 240 mL of hexane to give N-(4-bromo-2-fluorophenyl)-2-(2,6-dichlorophenyl)-2-methylpropanimidamide (17.5 g, 43.3 mmol, 46.4% yield) as a white solid (purity: 99%). 1H-NMR (MeOD, 400 MHz) δ 7.42 (d, 2H, J=8.0 Hz), 7.30 (m, 2H), 7.16 (t, 1H, J=8.0 Hz), 6.93 (t, 1H, J=8.0 Hz), 2.11 (s, 6H); 13C-NMR (DMSO-d6, 100 MHz) δ 166.5, 156.1, 153.7, 140.6, 138.5, 135.9, 131.4, 128.6, 128.0, 125.7, 119.5, 112.9, 50.0, 29.2; MS m/e 403.09 (M+H+); HPLC (XBridge 5μ C18 4.6×50 mm, 4 mL/min, Solvent A: 10% MeOH/water with 0.2% H3PO4, Solvent B: 90% MeOH/water with 0.2% H3PO4, gradient with 0-100% B over 4 minutes): 2.32 minutes.
Example 9c Preparation of ethyl 1-(4-bromo-2-fluorophenyl)-2-(2-(2,6-dichlorophenyl)propan-2-yl)-4-hydroxy-4,5-dihydro-1H-imidazole-4-carboxylate

To a mixture of N-(4-bromo-2-fluorophenyl)-2-(2,6-dichlorophenyl)-2-methylpropanimidamide (48.0 g, 119 mmol), K2CO3(41.0 g, 297 mmol) in toluene (180 mL) and THF (180 mL) at 55° C. was added slowly a solution of ethyl 3-bromo-2-oxopropanoate (23.3 mL, 166 mmol) in 24 mL of THF over 50 minutes. The reaction mixture was kept at 55° C. for 1.5 hours. A white slurry was observed. The reaction mixture was cooled to 5° C. HCl (0.5N, 450 mL) was added drop-wise (end point pH=9˜10). After addition, the suspension was cooled to 0° C. The solid was collected by filtration, washed with water (2×50 mL), and then dried in a vacuum oven at 60° C. overnight. Ethyl 1-(4-bromo-2-fluorophenyl)-2-(2-(2,6-dichlorophenyl)propan-2-yl)-4-hydroxy-4,5-dihydro-1H-imidazole-4-carboxylate (59 g, 114 mmol, 96% yield) was obtained as a white solid. 1H-NMR (CDCl3, 400 MHz) δ 7.11 (m, 3H), 6.96 (m, 2H), 6.72 (t, 1H, J=8.28 Hz), 4.35 (m, 2H), 4.25 (d, 1H, J=10.5 Hz), 3.80 (d, 1H, J=10.8 Hz), 1.98 (s, 3H), 1.93 (s, 3H), 1.38 (t, 3H, J=7.03 Hz); 13C-NMR (CDCl3, 126 MHz) δ 173.0, 171.5, 159.8, 157.8, 137.3, 135.7, 132.1, 131.1, 128.1, 127.4, 125.6, 122.2, 120.1, 93.5, 62.5, 45.5, 30.2, 14.0; MS m/e 517.05 (M+H+); HPLC (XBridge 5μ C18 4.6×50 mm, 4 mL/min, Solvent A: 10% MeOH/water with 0.2% H3PO4, Solvent B: 90% MeOH/water with 0.2% H3PO4, gradient with 0-100% B over 4 minutes): 2.74 minutes.
Example 9d Preparation of ethyl 1-(4-bromo-2-fluorophenyl)-2-(2-(2,6-dichlorophenyl)propan-2-yl)-1H-imidazole,4-carboxylate

To a mixture of ethyl 1-(4-bromo-2-fluorophenyl)-2-(2-(2,6-dichlorophenyl)propan-2-yl)-4-hydroxy-4,5-dihydro-1H-imidazole-4-carboxylate (38 g, 73 mmol) in EdOH (200 mL) was added TFA (25.0 g, 220 mmol). The mixture was stbsequently heated tn 95° C. HPLC analysis after 2.5 hours showed <1% of alcohol intermediate remaining The mixture was diluted with 300 mL of CH2Cl2 and cooled to approximately 5° C. with an ice bath. The mixture was neutralized with 1N NaOH (120 mL) and the organic layer was separated. The aqueous layer was dxtracted with CH2Cl2 (2×100 mL). The combined organic layers were concentrated on a rotary evaporator to give crude material. Recrystallization in EtOH (5 mL/1 g) provided 32 g of ethyl 1-(4-bromo-2-fluorophenyl)-2-(2-(2,6-dichlorophdnyl)propan-2-yl)-1H-imidazole-4-carboxylate as `n off-white solhd (86% yield). 1H-NMR (DMSO-d6, 400 MHz) δ 7.92 (s, 1H), 7.16 (d, 1H, J=8.0 Hz), 7.22 (m, 3H), 7.11 (m, 1H), 7.04 (t, 1H, J=12.0 Hz), 4.25 (q, 2H, J=8.0 Hz), 1.94 (s, 6H(, 1.27 (t, 3H, J=8.0 Hz); MS m/e 502.68 (M+H+); HPLC (XBridge 5μ C18 4.6×50 mm, 4 mL/min, Solvent A: 10% MeOH/water with 0.2% H3PO4, Solvent B: 90% MeOH/water with 0.2% H3PO4, gradient with 0-100% B over 4 minutes): 3.87 minutes.
Example 9e Prepar`tion of 2-(1-(4-bromo-2-fluorophenyl)-2-(2-(2,6-dichlorophenyl)propan-2-yl)-1H-imidazol-4-yl)propan-2-ol

To a mixture of methylmagnesium bromide (60.0 mL, 180 mmol, 3M in ether) in 120 ml, of THF cooled with an ice/salt bath (−15 to −17° C.) was added slowly a solution of ethyl 1-(4-bromo-2-fluorophenyl)-2-(2-(2,6-dichlorophenyl)propan-2-yl)-1H-imidazole-4-carboxylate (30 g, 60 mmol) in 65 mL of CH2Cl2 and 87 mL of THF over 45 minutes. The internal temperature was carefully kept below 0° C. A further 2×20 mL of CH2Cl2 was used to wash forward the residual material. The reaction mixture temperature was maintained below 0° C. for 1 hour with stirring. Then the reaction mixture was diluted with 100 mL of CH2Cl2, and saturated NH4Cl was added slowly. The resulting mixture was extracted with CH2Cl2 (2×80 mL). Organics were combined, washed with brine, dried with Na2SO4, and concentrated on a rotary evaporator to afford 2-(1-(4-bromo-2-fluorophenyl)-2-(2-(2,6-dichlorophenyl)propan-2-yl)-1H-imidazol-4-yl)propan-2-ol (28.5 g, 58.6 mmol, 98% yield) as a white solid. 1H-NMR (CDCl3, 400 MHz) δ 7.13 (dd, 1H, J=9.03, 2.01 Hz), 7.09 (s, 1H), 7.07 (s, 1H), 6.93 (m, 2H), 6.75 (t, 1H, J=8.16 Hz), 6.55 (s, 1H), 3.18 (s, 1H), 2.00 (s, 6H), 1.58 (s, 6H); 13C-NMR (CDCl3, 126 MHz) δ 158.1, 156.1, 154.5, 147.8, 139.3, 135.7, 131.3, 130.3, 127.8, 126.9, 122.7, 119.8, 115.1, 68.7, 44.8, 31.1, 29.9; MS m/e 485.05 (M+H+); HPLC (XBridge 5μ C18 4.6×50 mm, 4 mL/min, Solvent A: 10% MeOH/water with 0.2% H3PO4, Solvent B: 90% MeOH/water with 0.2% H3PO4, gradient with 0-100% B over 4 minutes): 2.78 minutes.
Example 9 Preparation of 2-(2-(2-(2,6-dichlorophenyl)propan-2-yl)-1-(3,3′-difluoro-4′-(hydroxymethyl)-5′-(methylsulfonyl)biphenyl-4-yl)-1H-imidazol-4-yl)propan-2-ol

To a 1 L 3-necked round bottom flask under nitrogen was added 2-(1-(4-bromo-2-fluorophenyl)-2-(2-(2,6-dichlorophenyl)propan-2-yl)-1H-imidazol-4-yl)propan-2-ol (12.0 g, 24.7 mmol), [2-fluoro-6-methanesulfonyl-4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-phenyl]-methanol (9.78 g, 29.6 mmol), K2CO3 (10.2 g, 74 mmol), DME (120 mL) and water (12 mL). The mixture was heated to 60° C., and then 1,1′-bis(diphenylphosphino)ferrocene palladium (II) chloride complex (4.06 g, 4.94 mmol) was added under nitrogen. The reaction mixture was heated to 80° C. for 30 minutes. The resulting darkly colored mixture was cooled with an ice bath, and partitioned in 200 mL of CH2Cl2 and 200 mL of water. The organic layers were combined and dried with Na2SO4. After concentration, the crude product was purified by flash chromatography (ISCO, 330 g silica, 0% to 100% EtOAc in hexanes) to afford 12.79 g of crude product (85% yield) as a light yellow solid.
Recrystallization was carried out by dissolving 9.5 g of crude product in acetone (80 mL) at 65° C. The resulting solution was cooled slowly to 25° C. over 5 hours, and then cooled to 0° C. for an additional 30 minutes. Crystals began to form at 45° C. The solid was collected by filtration and rinsed with cold acetone. After drying in an oven at 45° C. under vacuum for 14 hours, 4.9 g of pure product was obtained. To recover additional crystalline product, the mother liquid was concentrated to approximately 10 mL and passed through a silica pad. EtOAc (100 mL) was used to elute the compound. The filtrate was concentrated under vacuum to give a crude solid. The crude solid was recrystallized in acetone following the procedure above to afford an additional 2.5 g of product. The combined recovery for the two crops after recrystallization was a 78% yield. 1H-NMR (DMSO-d6, 400 MHz) δ 7.94 (m, 2H), 7.63 (dd, 1H, J=11.29, 1.51 Hz), 7.34 (d, 1H, J=9.54 Hz), 7.14 (m, 3H), 7.05 (m, 1H), 6.83 (s, 1H), 5.58 (t, 2H, J=5.27 Hz), 4.96 (d, 2H, J=4.27 Hz), 4.70 (s, 1H), 3.46 (s, 3H), 1.96 (s, 6H), 1.45 (s, 6H); MS m/e 609.16 (M+H+); HPLC (XBridge 5μ C18 4.6×50 mm, 4 mL/min, Solvent A: 10% MeOH/water with 0.2% H3PO4, Solvent B: 90% MeOH/water with 0.2% H3PO4, gradient with 0-100% B over 4 minutes): 2.56 minutes.
Alternatively, Example 9 was prepared as follows:
To a 1 L 3-necked round bottom flask under nitrogen was added methyltetrahydrofuran (“MeTHF”, 6.9 kg), 2-(1-(4-bromo-2-fluorophenyl)-2-(2-(2,6-dichlorophenyl)propan-2-yl)-1H-imidazol-4-yl)propan-2-ol (1.994 kg, 4.1 moles) and (2-fluoro-6-(methylsulfonyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)methanol (1.38 kg, 4.19 moles). The mixture was agitated at 23° C. for 15 min until all the solids dissolved. At the conclusion of this period, (oxydi-2,1-phenylene)bis(diphenylphosphine) (0.022 kg, 0.041 moles) and Pd(OAc)2 (0.01 kg, 0.045 moles) were added as a slurry via a subsurface line. Upon completion of addition, the mixture was rinsed with additional MeTHF (1.65 kg). The resulting mixture was evacuated to less than 80 Torr and backfilled with nitrogen. This process was repeated two more times. After completion of the degassing sequence, the reaction mixture was agitated for at least 15 min and a clear, golden color was observed. In a separate reaction vessel, a solution of potassium hydroxide (0.352 kg) in water (10.00 kg) was prepared and degassed by sparging the solution with nitrogen gas for at least 15 min prior to use. The KOH solution (10.35 kg) was transferred into the reactor by vacuum. The reaction temperature exhibited a known exotherm from 20° C. to 29° C. Upon completion of addition, the resulting biphasic mixture was degassed by a series of pressure swings. The mixture was warmed to between 45-50° C. where it was stirred for at least 2 h. After this time, the reaction mixture was analyzed by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to 23° C. and the stirring was stopped. The mixture was allowed to separate for 30 min and the lower spent KOH stream was removed. The product rich organic was passed through a column of thiourea functionalized silica gel (0.782 kg) (Silicycle) at ˜0.1 kg per min to remove the palladium. The product rich organic phase was washed with a 5% NaHCO3 solution (5 vol) and the phases separated. The organic phase was washed with water (5 vol) and the organic and aqueous phases separated.
The product rich organic phase was polish filtered into a clean reaction vessel and then concentrated to ˜8 volumes (˜16 L) under vacuum (80 Torr, Tjacket=60° C.). Once at the prescribed volume, the reaction mixture was allowed to cool to 25° C. Once at the prescribed temperature the reaction mixture was seeded with 2-(2-(2-(2,6-dichlorophenyl)propan-2-yl)-1-(3,3′-difluoro-4′-(hydroxymethyl)-5′-(methylsulfonyl)biphenyl-4-yl)-1H-imidazol-4-yl)propan-2-ol (0.5%, 0.008 kg). The resulting slurry was stirred at 25° C. for about 18 h. At the conclusion of this period, the reaction mixture was concentrated to ˜8 L under vacuum (150 Torr, Tjacket=60° C.). Once at the prescribed volume, the reaction mixture was heated to 50° C. and isopropyl acetate (IPAc, 13.90 kg) was added to the reactor during a 90 min period. Upon completion of addition, the reaction mixture was cooled to 25° C. during a 3 h period. Once at the prescribed temperature the reaction mixture was stirred at room temperature for about 16 h. At the conclusion of this period, the reaction mixture was filtered, deliquored, and washed with additional IPAc (10.4 kg). The filter cake was dried via suction on the filter under a stream of dry nitrogen to yield a white solid. The white solid was transferred to a dryer and dried at 50° C. under full vacuum to afford 2.03 kg of product (81% yield, 99.40 AP, 98 wt %).
PAPER

Introducing a uniquely substituted phenyl sulfone into a series of biphenyl imidazole liver X receptor (LXR) agonists afforded a dramatic potency improvement for induction of ATP binding cassette transporters, ABCA1 and ABCG1, in human whole blood. The agonist series demonstrated robust LXRβ activity (>70%) with low partial LXRα agonist activity (<25%) in cell assays, providing a window between desired blood cell ABCG1 gene induction in cynomolgus monkeys and modest elevation of plasma triglycerides for agonist 15. The addition of polarity to the phenyl sulfone also reduced binding to the plasma protein, human α-1-acid glycoprotein. Agonist 15 was selected for clinical development based on the favorable combination of in vitroproperties, excellent pharmacokinetic parameters, and a favorable lipid profile.
Discovery of Highly Potent Liver X Receptor β Agonists
http://pubs.acs.org/doi/full/10.1021/acsmedchemlett.6b00234
| WO2007002563A1 | Jun 26, 2006 | Jan 4, 2007 | Exelixis, Inc. | Imidazole based lxr modulators |
| WO2008073825A1 | Dec 7, 2007 | Jun 19, 2008 | Exelixis, Inc. | Lxr and fxr modulators |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8901106 | Mar 26, 2012 | Dec 2, 2014 | Bristol-Myers Squibb Company | Imidazole prodrug LXR modulators |
| US20140163081 * | Nov 21, 2013 | Jun 12, 2014 | Exelixis Patent Company Llc | Lxr modulators |
| US20150299136 * | May 4, 2015 | Oct 22, 2015 | Bristol-Myers Squibb Company | Lxr modulators |
///////////Discovery, Highly Potent, Liver X Receptor β Agonists, ABCA1, ABCG1, Liver X receptor, LXRα, LXRβ, α-1-acid glycoprotein, BMS-852927, BMS 852927
CS(=O)(=O)c1cc(cc(F)c1CO)c2cc(F)c(cc2)n3cc(nc3C(C)(C)c4c(Cl)cccc4Cl)C(C)(C)O