














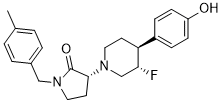
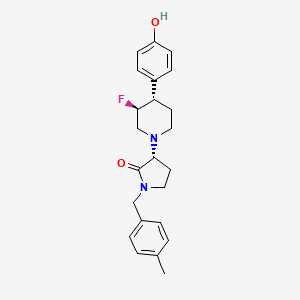
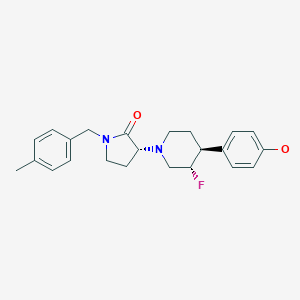
BMS-986169
CAS 1801151-08-5 Related CAS : 1801151-09-6 1801151-08-5
Chemical Formula: C23H27FN2O2
Molecular Weight: 382.4794
Elemental Analysis: C, 72.23; H, 7.12; F, 4.97; N, 7.32; O, 8.37
(R)-3-((3S,4S)-3-fluoro-4-(4-hydroxyphenyl)piperidin-1-yl)-1-(4-methylbenzyl)pyrrolidin-2-one
(3R)-3-[(3S,4S)-3-fluoro-4-(4-hydroxyphenyl)piperidin-1-yl]-1-[(4-methylphenyl)methyl]pyrrolidin-2-one
Preclinical
BMS-986169 is a Novel, Intravenous, Glutamate N-Methyl-d-Aspartate 2B Receptor Negative Allosteric Modulator with Potential in Major Depressive Disorder. BMS-986169 showed high binding affinity for the GluN2B subunit allosteric modulatory site (Ki = 4.03-6.3 nM) and selectively inhibited GluN2B receptor function in Xenopus oocytes expressing human N-methyl-d-aspartate receptor subtypes (IC50 = 24.1 nM). BMS-986169 weakly inhibited human ether-a-go-go-related gene channel activity (IC50 = 28.4 μM) and had negligible activity in an assay panel containing 40 additional pharmacological targets.
Chemical structures of BMS-986169 and the phosphate prodrug BMS-986163.

PAPER
Evolution of a Scale-Up Synthesis to a Potent GluN2B Inhibitor and Its Prodrug
 , Huiping Zhang†, Michael K. Y. Wong†, Jianqing Li† , Peng Li†, Dauh-Rurng Wu†, Richard Rampulla†, Michael A. Galella‡, Marta Dabros‡, Sarah C. Traeger†, Vetrichelvan Muthalagu§, Anuradha Gupta§, Pirama Nayagam Arunachalam§, and Arvind Mathur†
, Huiping Zhang†, Michael K. Y. Wong†, Jianqing Li† , Peng Li†, Dauh-Rurng Wu†, Richard Rampulla†, Michael A. Galella‡, Marta Dabros‡, Sarah C. Traeger†, Vetrichelvan Muthalagu§, Anuradha Gupta§, Pirama Nayagam Arunachalam§, and Arvind Mathur†
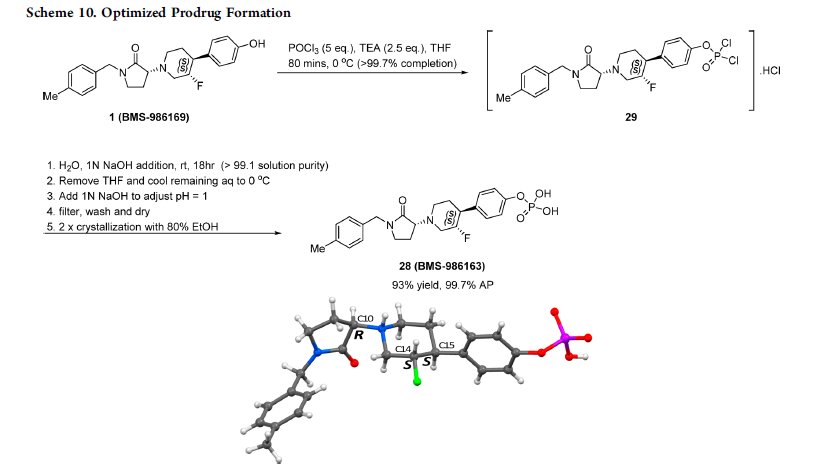
This paper describes the efficient scale-up synthesis of the potent negative allosteric glutamate N2B (GluN2B) inhibitor 1 (BMS-986169), which relies upon a stereospecific SN2 alkylation strategy and a robust process for the preparation of its phosphate prodrug 28 (BMS-986163) from parent 1 using POCl3. A deoxyfluorination reaction employing bis(2-methoxyethyl)aminosulfur trifluoride (Deoxo-Fluor) is also used to stereospecifically introduce a fluorine substituent. The optimized routes have been demonstrated to provide APIs suitable for toxicological studies in vivo.
https://pubs.acs.org/doi/suppl/10.1021/acs.oprd.8b00120/suppl_file/op8b00120_si_001.pdf
PAPER
https://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.8b00080
BMS-986163, a Negative Allosteric Modulator of GluN2B with Potential Utility in Major Depressive Disorder
, ET AL

There is a significant unmet medical need for more efficacious and rapidly acting antidepressants. Toward this end, negative allosteric modulators of the N-methyl-d-aspartate receptor subtype GluN2B have demonstrated encouraging therapeutic potential. We report herein the discovery and preclinical profile of a water-soluble intravenous prodrug BMS-986163 (6) and its active parent molecule BMS-986169 (5), which demonstrated high binding affinity for the GluN2B allosteric site (Ki = 4.0 nM) and selective inhibition of GluN2B receptor function (IC50 = 24 nM) in cells. The conversion of prodrug 6 to parent 5 was rapid in vitro and in vivo across preclinical species. After intravenous administration, compounds 5 and 6 have exhibited robust levels of ex vivo GluN2B target engagement in rodents and antidepressant-like activity in mice. No significant off-target activity was observed for 5, 6, or the major circulating metabolites met-1 and met-2. The prodrug BMS-986163 (6) has demonstrated an acceptable safety and toxicology profile and was selected as a preclinical candidate for further evaluation in major depressive disorder.

(S)-3-((3S,4S)-3-fluoro-4-(4-hydroxyphenyl)piperidin-1-yl)-1-(4-
methylbenzyl)pyrrolidin-2-one (compound 23) and (R)-3-((3S,4S)-3-fluoro-4-(4-
hydroxyphenyl)piperidin-1-yl)-1-(4-methylbenzyl)pyrrolidin-2-one (BMS-986169, compound
5)……https://pubs.acs.org/doi/suppl/10.1021/acsmedchemlett.8b00080/suppl_file/ml8b00080_si_001.pdf
Analytical data for BMS-986169 (compound 5): LCMS (C23H27FN2O2, MW 382.2, ESAPI),
observed 383.2 m/z (M+H)+; []D20 = +6.09 (c = 1.15, MeOH); Anal. Calcd for
C23H27FN2O2 (382.21): C, 72.22; H, 7.12; N, 7.32. Found: C, 72.26; H, 7.05; N, 7.31; HRMS
(ESI) Calcd for C23H27N2O2, 383.2118. Found, 383.2129;
13C NMR (126 MHz, chloroformd)
172.4, 155.0, 137.5, 133.0, 132.8, 129.4, 128.6, 128.2, 115.6, 91.6 (d, J=173.5 Hz),
65.0, 54.5 (d, J=25.4 Hz), 48.3, 47.7 (d, J=17.3 Hz), 46.7, 43.6, 31.5, 21.1, 19.2;(500 MHz, chloroform-d) 7.23 – 7.11 (m, 5H), 6.92 (d, J=8.5 Hz, 2H), 6.18 (br. s., 1H),
4.79 – 4.55 (m, 1H), 4.57 – 4.33 (m, 2H), 3.72 (t, J=8.7 Hz, 1H), 3.46 – 3.30 (m, 1H), 3.30 –
3.09 (m, 2H), 2.82 (d, J=8.5 Hz, 1H), 2.73 – 2.56 (m, 2H), 2.49 (d, J=2.5 Hz, 1H), 2.36 (s,
3H), 2.21 – 1.98 (m, 2H), 1.87 (br. s., 2H). The corresponding 1H NMR spectrum for
compound 5 is shown below
1H NMR

PATENT
https://patents.google.com/patent/US9221796B2/und
InventorDalton KingLorin A. Thompson, IIIJianliang ShiSrinivasan ThangathirupathyJayakumar Sankara WarrierImadul IslamJohn E. Macor
Current Assignee Bristol-Myers Squibb Co
https://patents.google.com/patent/WO2015105772A1/und
N-Methyl-D-aspartate (NMDA) receptors are ion channels which are gated by the binding of glutamate, an excitatory neurotransmitter in the central nervous system. They are thought to play a key role in the development of a number of neurological diseases, including depression, neuropathic pain, Alzheimer’s disease, and Parkinson’s disease. Functional NMDA receptors are tetrameric structures primarily composed of two NRl and two NR2 subunits. The NR2 subunit is further subdivided into four individual subtypes: NR2A, NR2B, NR2C, and NR2D, which are differentially distributed throughout the brain. Antagonists or allosteric modulators of NMDA receptors, in particular NR2B subunit-containing channels, have been investigated as therapeutic agents for the treatment of major depressive disorder (G. Sanacora, 2008, Nature Rev. Drug Disc. 7: 426-437).
The NR2B receptor contains additional ligand binding sites in additon to that for glutamate. Non-selective NMDA antagonists such as Ketamine are pore blockers, interfering with the transport of Ca++ through the channel. Ketamine has demonstrated rapid and enduring antidepressant properties in human clinical trials as an i.v. drug. Additionally, efficacy was maintained with repeated, intermittent infusions of Ketamine (Zarate et al., 2006, Arch. Gen. Psychiatry 63: 856-864). This class of drugs, though, has limited therapeutic value because of its CNS side effects, including dissociative effects.
An allosteric, non-competitive binding site has also been identified in the N-terminal domain of NR2B. Agents which bind selectively at this site, such as
Traxoprodil, exhibited a sustained antidepressant response and improved side effect profile in human clinical trials as an i.v. drug (Preskorn et al., 2008, J. Clin.
PsychopharmacoL, 28: 631-637, and F. S. Menniti, et al, 1998, CNS Drug Reviews, 4, 4, 307-322). However, development of drugs from this class has been hindered by low bioavailability, poor pharmacokinetics, and lack of selectivity against other pharmacological targets including the hERG ion channel. Blockade of the hERG ion channel can lead to cardiac arrythmias, including the potentially fatal Torsades de pointe, thus selectivity against this channel is critical. Thus, in the treatment of major depressive disorder, there remains an unmet clinical need for the development of effective NR2B-selective negative allosteric modulators which have a favorable tolerability profile.
NR2B receptor antagonists have been disclosed in PCT publication WO 2009/006437.
The invention provides technical advantages, for example, the compounds are novel and are ligands for the NR2B receptor and may be useful for the treatment of various disorders of the central nervous system. Additionally, the compounds provide advantages for pharmaceutical uses, for example, with regard to one or more of their mechanism of action, binding, inhibition efficacy, target selectivity, solubility, safety profiles, or bioavailability.
Synthetic Scheme 1

The l-phenyl/benzyl-3-bromo-pyrrolidinones/piperidinones V may be reacted with (4-oxy-phenyl)cyclic amines VI in the presence of base to produce protected products VII, which may be subjected to cleavage conditions appropriate for the protecting group (PGi) to generate final products I, which may be separated into individual enantiomers/diastereomers I*, as shown in synthetic scheme 2.
Synthetic Scheme 2

I I*
Compounds la may be prepared by condensing l-phenyl/benzyl-3-bromo-pyrroli-dinones/piperidinones V with substituted 4(4-oxyphenyl)piperidines Vllla-c to generate protected intermediates IX, which may be subjected to cleavage conditions appropriate for the protecting group (PGi) to generate final products la, which may be separated into individual enantiomers/diastereomers la*, as shown in synthetic scheme 3.
Synthetic Scheme 3

The 4(4-oxyphenyl)piperidines Vllla-c may be synthesized in turn by a sequence starting with a protected tetrahydropiperidine X, which can be hydroxylated via hydroboration/oxidation to give the protected hydroxypiperidine XI, which may be either directly transformed into the protected fluoropiperidine XII by treatment with DAST or oxidized into the protected 3-oxopiperidine XIII, which may be further transformed into protected 3,3-difluoropiperidines XIV via treatment with DAST. XI, XII, and XIV may be transformed into Villa, Vlllb, and VIIIc, respectively, by employing cleaving conditions appropriate for the protecting group (PG2), as shown in synthetic scheme 3 a.
S nthetic scheme 3 a
![]()
![]()
Chiral
Cleavage Individual enantiomers/
G2P-N diastereomers
separation
conditions
OH ![]()
Villa*
Villa
XI
Chiral
Cleavage Individual enantiomers/
HN
G2P-N diastereomers
%_\J> PQ separation
PG1 conditions
R F
F Vlllb*
Vlllb
XII
Chiral
Individual enantiomers/
G2P- diastereomers separation
Vlllc*

For tetrahydropyridines X which are not commercially available may be synthesized by coupling protected bromophenols XV with protected unsaturated
piperidineboronic acids XVI, as shown in synthetic scheme 4a.
Synthetic scheme 4a:

For tetrahydropyridines X which are not commercially available may be synthesized by adding the anion generated from protected bromophenols XV to a protected 4-piperidinone XVII to yield 4-phenyl-4-piperidinol XVIII, which may be dehydrated under acid conditions to yield the desired X, as shown in synthetic scheme 4b.
Synthetic scheme 4b:

l-Phenyl/benzyl-3-bromo-pyrroli-dinones/piperidinones V may be condensed with isolated individual enantiomers VIIIa-c*, which results in diastereomers 1- phenyl/benzyl-3-bromo-pyrroli-dinones/piperidinones IX*, which may be deprotected and separated to give final products la*, as shown in scheme 5.

Alternatively, the backbone scaffold may be synthesized by condensing 1- phenyl/benzyl-3-bromo-pyrroli-dinones/piperidinones V with hydroxypiperidines Villa to yield the protected 3-fluoropiperidines IXa, which may themselves be converted to the protected 3-fluoropiperidines IXb or oxidized to the ketones XIX, which may be converted to the 3,3-difluoropiperidines Ixc, as shown in scheme 6. The final compounds can then be isolated after the deprotection of IXa-c.
Scheme 6

Example 46, P-1 Example 46, P-2
(S)-3-((3S,4S)-3-Fluoro-4-(4-hydroxyphenyl)piperidin-l-yl)-l-(4-methylbenzyl)pyrrolidin-2-one and (R)-3-((3S,4S)-3-fluoro-4-(4-hydroxyphenyl)piperidin-l-yl)-l-(4-methylbenzyl)pyrrolidin-2-one.

Example 46, P-3 Example 46, P-4
Step A. (±)-rel-(3S,4S)- 1 -benzyl-4-(4-methoxyphenyl)piperidin-3-ol.

To a suspension of sodium tetrahydroborate (2.7 g, 72 mmol) in THF (200 mL) at 0 °C under a nitrogen atmosphere was added dropwise boron trifluoride etherate (8.8 mL, 70 mmol) and the resulting mixture was stirred for 30 minutes. Then 1-benzyl- 4-(4-methoxyphenyl)-l,2,3,6-tetrahydropyridine (10 g, 36 mmol, from S. Halazy et al WO 97/28140 (8/7/97)) dissolved in 100 mL of tetrahydrofuran was added. The mixture was allowed to warm to rt and stirred for 2 h. The reaction was then quenched by the dropwise addition of 100 mL of water. Next were added
sequentially 100 mL of ethanol, 100 mL of a 10% aqueous sodium hydroxide solution, and 30%> hydrogen peroxide (18 mL, 180 mmol) and the mixture was stirred at reflux temperature overnight. The reaction mixture was then allowed to cool, diluted with saturated aqueous ammonium chloride (200 mL), and extracted with ethyl acetate (500 mL). The organic layer was dried over Na2S04, filtered, and evaporated under reduced pressure to give (±)-rel-(3S,4S)- 1 -benzyl-4-(4-methoxyphenyl)piperidin-3-ol (8.5 g, 24.6 mmol, 69%> yield) which was used without further purification. LCMS (Method K) RT 1.99 min; m/z 298.0 (M+H+).
Step B. (±)-re -(3S,4S)-4-(4-methoxyphenyl)piperidin-3-ol.

To a solution of (±)-re/-(35′,45)-l-benzyl-4-(4-methoxyphenyl)piperidin-3-ol (9 g, 30 mmol) in methanol (150 mL) was added 10 % Pd/C (4.8 g) and the reaction mixture was stirred overnight under a hydrogen atmosphere. The catalyst was then removed by filtration through Celite and the solvent was evaporated under reduced pressure to give (±)-re/-(3S,4S)-4-(4-methoxyphenyl)piperidin-3-ol (5.1 g, 24.6 mmol, 81% yield) which was used without further purification. 1H NMR (400 MHz, DMSO-de) δ ppm 7.10 – 7.15 (m, 2 H) 6.80 – 6.86 (m, 2 H) 4.30 (d, J=5.27 Hz, 1 H) 3.37 – 3.43 (m, 1 H) 3.04 (dd, J=11.58, 4.36 Hz, 1 H) 2.86 (d, J=12.17 Hz, 1 H) 2.43 (td, J=12.09, 2.67 Hz, 1 H) 2.22 – 2.35 (m, 2 H) 1.57 – 1.63 (m, 1 H) 1.43 – 1.54 (m, 1 H).

To a solution of (±)-re/-(3S,4S)-4-(4-methoxyphenyl)piperidin-3-ol (4.5 g, 21.7 mmol) in DCM (150 mL) at -10°C under nitrogen was added a 1 M solution of boron tribromide in DCM (109 mL, 109 mmol). The reaction mixture was allowed to warm to rt, stired for 2 h, and then rechilled to 0 °C and quenched by the addition of a saturated aqueous sodium bicarbonate solution (300 mL). The aqueous layer was washed with 250 mL of DCM and then to it was added 200 mL 10% aqueous NaOH, followed by 9.5 g (43.5 mmol) of di-t-butyl dicarbonate and the resulting mixture was stirred for an additional 2 h. The mixture was then extracted with 200 mL ethyl acetate and the organic layer was separated, dried over Na2S04,filtered, and evaporated under reduced pressure to (±)-re/-(35′,45)-tert-butyl 4-(4-(tert-butoxycarbonyloxy)phenyl)-3-hydroxypiperidine-l-carboxylate (6.5 g, 12 mmol, 56 % yield) which was used without further purification. LCMS (Method K) RT 2.33 min, m/z 282 (M+H+ -2 t-butyl), 370; 1H NMR (400 MHz, DMSO-d6) δ ppm 7.27 (d, J=8.66 Hz, 2 H) 7.08 (d, J=8.66 Hz, 2 H) 4.85 (d, J=5.65 Hz, 1 H) 4.13 (d, J=8.41 Hz, 1 H) 3.97 (d, J=10.48 Hz, 1 H) 3.45 (tt, J=10.27, 5.19 Hz, 1 H) 1.67 (d, J=3.39 Hz, 1 H) 1.50 – 1.59 (m, 1 H) 1.49 (s, 11 H).
Step D. (±)-re/-(35′,45)-tert-butyl 3-hydroxy-4-(4-hydroxyphenyl)piperidine-l-carboxylate.

To a solution of (±)-re/-(35′,45)-tert-butyl 4-(4-(tert-butoxycarbonyloxy)phenyl)-3-hydroxypiperidine-l-carboxylate (6.5 g, 16.5 mmol) in 100 mL of methanol was added 11.42 g of potassium carbonate (83 mmol) and the reaction mixture was stirred at rt for 5 h. The organic solvent was removed under reduced pressure and the residue was partitioned between IN HC1 (300 mL) and ethyl acetate (300 mL). The layers were separated and the organic layer was dried over Na2S04 and evaporated under reduced pressure to give (±)-re/-(35′,45)-tert-butyl 3-hydroxy-4-(4-hydroxyphenyl)piperidine-l-carboxylate (5 g, 15 mmol, 92 % yield) which was used without further purification. LCMS (method F) RT 1.85 min, m/z 238 (M+H+ – 1-butyl), 279 (M+H+ – t-butyl+CH3CN), 1H NMR (400 MHz, DMSO-d6) δ ppm 7.01 (d, J=8.53 Hz, 2 H) 6.66 (d, J=8.53 Hz, 2 H) 4.70 (d, J=5.02 Hz, 1 H) 4.09 (br. s., 1 H) 3.94 (d, J=11.55 Hz, 1 H) 3.35 – 3.41 (m, 1 H) 2.66 – 2.77 (m, 1 H) 2.29 – 2.39 (m, 1 H) 1.63 (dd, J=13.30, 3.26 Hz, 1 H) 1.44 – 1.52 (m, 1 H) 1.42 (s, 9 H).
Step E. (3S,4S)-tert-Butyl 3 -hydroxy-4-(4-hydroxyphenyl)piperidine-l -carboxylate and (3R, -tert-butyl 3-hydroxy-4-(4-hydroxyphenyl)piperidine-l-carboxylate.

E-1 E-2
(±)-rel-(3S,4S)-tert-Butyl 3-hydroxy-4-(4-hydroxyphenyl)piperidine- 1 -carboxylate (5 g, 17 mmol, from step D) was subjected to chiral SFC separation (method C-5) to yield enantiomers E-1 (1.9 g, 6.48 mmol, 38.0 % yield) and E-2 (2.4 g, 8.18 mmol, 48.0 % yield). Data for E-1 : chiral HPLC (method A5 ) retention time 3.42 min. Data for E-2: chiral HPLC (method A5) retention time 4.2 min.
Step F. (3R,4R)-tert-Butyl 4-(4-(benzyloxy)phenyl)-3-hydroxypiperidine-l-carboxylate.

A mixture of (3R,4R)-tert-butyl 3-hydroxy-4-(4-hydroxyphenyl)piperidine-l-carboxylate (620 mg, 2.1 mmol, E-2 from step E), potassium carbonate (584 mg, 4.2 mmol), and benzyl bromide (0.25 mL, 2.1 mmol) in DMF (5 mL) was stirred at rt for 16 h. The solvent was removed by evaporation and the residue was treated with 50 mL of water. The aqueous mixture was then extracted 4 times with 50 mL of chloroform. The combined organic phases were dried over anhydous Na2S04, filtered, and evaporated to yield 750 mg of (3R,4R)-tert-butyl 4-(4-(benzyloxy)phenyl)-3-hydroxypiperidine-l -carboxylate which was used without further purification. LCMS (method F) RT 2.28 min, m/z = 310 (M+H+ – t-butyl -water), 328 (M+H+ -t-butyl).
Step G. (3i?,4i?)-4-(4-(Benzyloxy)phenyl)piperidin-3-ol hydrochloride.

A mixture of (3R,4R)-tert-butyl 4-(4-(benzyloxy)phenyl)-3-hydroxypiperidine-1-carboxylate (750 mg, 2 mmol), dioxane (4 mL) and 4.9 mL of 4 M HCI in dioxane was stirred at rt for 2h. The reaction was then evaporated to dryness to yield 550 mg of (3i?,4i?)-4-(4-(Benzyloxy)phenyl)piperidin-3-ol hydrochloride which was used without further purification. LCMS (method J) RT 0.70 min, m/z 284 (M+H+).
Step H. 3-((3i?,4i?)-4-(4-(Benzyloxy)phenyl)-3-hydroxypiperidin-l -yl)- 1 -(4-methylbenzyl)pyrrolidin-2-one .

A mixture of 3-bromo-l-(4-methylbenzyl)pyrrolidin-2-one (Intermediate 2, 220 mg, 0.82 mmol), (3i?,4i?)-4-(4-(benzyloxy)phenyl)piperidin-3-ol hydrochloride (262 mg, 0.82 mmol, from step G) and triethylamine (11 mL, 8.2 mmol) was stirred at 60 °C for lh, 80 °C for 1 h, 100 °C for 1 h and 120 °C for 1 h. The reaction mixture was then allowed to cool, diluted with 40 mL of water and extracted four times with 50 mL of chloroform. The combined organic layers were washed with 60 mL brine, dried over anhydrous sodium sulfate, filtered, and evaporated to yield 382 mg of 3-((3 ?,4i?)-4-(4-(benzyloxy)phenyl)-3-hydroxypiperidin- 1 -yl)- 1 -(4-methylbenzyl)pyrrolidin-2-one which was used without further purification. LCMS (method J) (main component of a mixture) RT 2.23 min, m/z 471 (M+H+).
Step I. 3-((3R, 4R)-4-(4-(Benzyloxy)phenyl)-3-fluoropiperidin- 1 -yl)- 1 -(4-methylbenzyl)pyrrolidin-2-one .

A solution of 3-(-4-(4-(benzyloxy)phenyl)-3-hydroxypiperidin-l-yl)-l-(4-methylbenzyl)pyrrolidin-2-one (382 mg, 0.81 mmol) in DCM (5 mL) cooled to 0 °C was treated dropwise with DAST (0.32 mL, 2.4 mmol) over 3 min. The reaction mixture was then allowed to warm to rt and was stirred for 2 h. The reaction was then quenched with 50 mL of 10% aqueous sodium bicarbonate solution and extracted 4 times with 40 mL of DCM. The combined organic layers were washed with 50 mL of brine, dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum to yield 382 mg of 3-((3i?,4i?)-4-(4-(benzyloxy)phenyl)-3-fluoropiperidin-l-yl)-l-(4-methylbenzyl)pyrrolidin-2-one as a mixture of two diastereomers and rearrangement products which was used without further purification. LCMS (method J) (main component of a mixture) RT 0.9 min, m/z 473 (M+H+).
Step J. 3-((3i?,4i?)-3-Fluoro-4-(4-hydroxyphenyl)piperidin- 1 -yl)-l -(4-methylbenzyl)pyrrolidin-2-one .

A mixture of 3-((Ji?,4i?)-(4-(4-(benzyloxy)phenyl)-3-fluoropiperidin-l-yl)-l-(4-methylbenzyl)pyrrolidin-2-one (382 mg, 0.81 mmol) and methanol (4 mL) was flushed with nitrogen, followed by the addition of 172 mg of 10% Pd/C. Then the mixture was stirred at rt overnight under 25-99 psi hydrogen pressure. The reaction was then transferred to a 100 mL autoclave and stirred at 7 kg/cm2 hydrogen pressure for 4 days. The catalyst was removed by filtration through Celite and the solvent was evaporated off. The crude product was subjected to HPLC purification (method B) to yield 77.3 mg 3-((Ji?,4i?)-3-fluoro-4-(4-hydroxyphenyl)-piperidin-l-yl)-l-(4-methylbenzyl)pyrrolidin-2-one (diastereomeric pair) LCMS (method Q) RT 1.15 min, m/z 383.0 (M+H+).
Step K. (5)-3-((3i?,4i?)-3-Fluoro-4-(4-hydroxyphenyl)piperidin- 1 -yl>
methylbenzyl)pyrrolidin-2-one and (i?)-3-((3i?,4i?)-3-fluoro-4-(4-hydroxyphenyl)piperidin- 1 -yl)- 1 -(4-methylbenzyl)pyrrolidin-2-one.
The diastereomeric mixture from step J was separated by SFC method C-7 to yield homochiral Examples 46 P-l (29.3 mg) and P-2 (32.8 mg). Data for P-l (S)-3-((3R, 4R)-3 -fluoro-4-(4-hydroxyphenyl)piperidin- 1 -yl)- 1 -(4-methylbenzyl)pyrrolidin-2-one: LCMS (method F) RT 2.10 min, m/z 383.2 (M+H+), 405.2 (M+Na+); HPLC (method B) RT 8.24 min (98.8% AP); HPLC (method C) RT 6.52 min (99.1% AP); Chiral HPLC (method C-6) RT 4.1 min; 1H NMR (400 MHz, methanol-d4) δ ppm 1.76 – 1.86 (m, 2 H) 2.07 (d, J=8.53 Hz, 1 H) 2.13 – 2.21 (m, 1 H) 2.34 (s, 3 H) 2.43 (s, 0 H) 2.55 – 2.60 (m, 1 H) 2.65 – 2.70 (m, 1 H) 2.75 (br. s., 1 H) 3.20 – 3.30 (m, 2 H) 3.38 – 3.45 (m, 1 H) 3.70 (t, J=8.78 Hz, 1 H) 4.44 (t, J=79.81 Hz, 3 H) 4.63 – 4.71 (m, 1 H) 6.70 – 6.80 (m, 2 H) 7.07 – 7.15 (m, 2 H) 7.07 – 7.12 (m, 1 H) 7.13 – 7.22 (m, 4 H); 19F NMR δ ppm -184.171. Data for P-2: (R)-3-((3R,4R)-3-fluoro-4-(4-hydroxyphenyl)piperidin- 1 -yl)- 1 -(4-methylbenzyl)pyrrolidin-2-one: LCMS (method F) RT 2.10 min, m/z 383.2 (M+H+), 405.2 (M+Na+); HPLC (method B) RT 8.29 min (99.7% AP); HPLC (method C) RT 6.52 min (99.8% AP); Chiral HPLC (method C-6) RT 6.92 min; 1H NMR (400 MHz, methanol-d4) δ ppm 1.80 – 1.90 (m, 2 H) 2.07 (d, J=8.03 Hz, 1 H) 2.19 (s, 1 H) 2.34 (s, 3 H) 2.41 – 2.48 (m, 1 H) 2.66 (d, J=4.52 Hz, 2 H) 2.95 – 3.03 (m, 1 H) 3.10 – 3.18 (m, 1 H) 3.20 – 3.30 (m, 2 H) 3.68 – 3.78 (m, 1 H) 4.38 (s, 1 H) 4.51 (d, J=14.56 Hz, 2 H) 6.70 – 6.80 (m, 2 H) 7.05 – 7.13 (m, 2 H) 7.13 – 7.22 (m, 4 H); 19F NMR δ ppm -184.311.
(3S,4S)-tert-Butyl 3-fluoro-4-(4-hydroxyphenyl)piperidine-l-carboxylate.

To a solution of (3S,4S)-tert-butyl 3-hydroxy-4-(4-hydroxyphenyl)piperidine-l-carboxylate (400 mg, 1.36 mmol, the first eluting enantiomer E-l from step E) in DCM (5 mL) cooled to 0 °C was added dropwise DAST (0.54 mL, 4.1 mmol) over 10 min. The mixture was allowed to warm up to rt and was stirred for 2h. The reaction was slowly quenched with 50 mL of a 10%> aqueous sodium bicarbonate solution and extracted four times with 50 mL of DCM. The combined organic layerss were washed with 75 mL of brine, dried, and concentrated under vacuum to yield 390 mg of {3S,4S)-tert-bvXy\ 3-fluoro-4-(4-hydroxyphenyl)piperidine-l-
carboxylate which was used without further purification. LCMS (Method Q) RT 0.92 min, m z 240.1(M+H+).
Step M. 4-((3S’,4S)-3-Fluoropi ridin-4-yl)phenol hydrochloride.

A mixture of (3S,4S)-tert-butyl 3-fluoro-4-(4-hydroxyphenyl)piperidine-l-carboxylate (390 mg, 1.3 mmol) and 4M HC1 in dioxane (3.3 mL, 13.2 mmol) in dioxane (4 mL) was stirred at rt for 2 hr. It was then concentrated to dryness, washed with 10 mL of 5% DCM/diethyl ether mixture and the solid was isolated by filtration. Yield: 260 mg of 4-((J£4S)-3-fluoropiperidin-4-yl)phenol hydrochloride; LCMS
(method Q) RT 0.46 min, mz 196.1(M+H+) 1H NMR (400 MHz, DMSO-d6) δ = 9.57 (br. s., 4 H), 8.92 – 8.68 (m, 1 H), 7.14 (d, J= 8.5 Hz, 1 H), 7.06 (d, J= 8.5 Hz, 2 H), 6.82 – 6.73 (m, 2 H), 5.07 – 4.85 (m, 1 H), 3.77 – 3.36 (m, 9 H), 3.32 – 3.22 (m, 2 H), 3.13 – 2.85 (m, 5 H), 2.06 – 1.88 (m, H).
Step N. 3-((3S,4S)-3-Fluoro-4-(4-hydroxyphenyl)piperidin- 1 -yl)- 1 -(4-methylbenzyl)pyrrolidin-2-one.

A mixture of 3-bromo-l-(4-methylbenzyl)pyrrolidin-2-one (200 mg, 0.75 mmol), triethylamine (0.52 mL, 3.7 mmol) and 4-((3S,4S)-3-fluoropiperidin-4-yl)phenol hydrochloride (173 mg, 0.75 mmol) in DMF (3 mL) was heated to 120 °C in a microwave reactor for 1.5 h. The mixture was allowed to cool and was then mixed with 60 mL water and extracted 5 times with 40 mL of DCM. The combined organic extracts were washed with 80 mL of brine, dried over anhydrous sodium sulfate, filtered, and evaporated to give 265 mg of 3-((3 4S)-3-fluoro-4-(4-hydroxy-phenyl)piperidin-l-yl)-l-(4-methylbenzyl)pyrrolidin-2-one as a mixture of 2 diastereoisomers. LCMS (method P) RT 0.92 min m/z 383.4 (M+H+).
Step O. (5)-3-((3lS,45)-3-Fluoro-4-(4-hydroxyphenyl)piperidin- 1 -yl)- 1 -(4-methylbenzyl)pyrrolidin-2-one and (i?)-3-((35,,45)-3-fluoro-4-(4-hydroxyphenyl)piperidin- 1 -yl)- 1 -(4-methylbenzyl)pyrrolidin-2-one.
A portion of the diasteromer mixture from step N (130 mg) was subjected to chiral purification via SFC (method C-7) to give homochiral Examples 46 P-3 (37.7 mg) and P-4 (60.7 mg). Data for P-3 (S)-3-((3S,4S)-3-fluoro-4-(4-hydroxyphenyl)piperidin- 1 -yl)- 1 -(4-methylbenzyl)pyrrolidin-2-one: LCMS (Method F) RT = 2.10 min, m/z 383.2 (M+H+); HPLC (Method C) RT 6.54 min, (Method D) RT 8.20 min; chiral HPLC (method C-6) RT 3.42 min;1H NMR (400 MHz, methanol-d4) δ ppm 1.76 – 1.86 (m, 2 H) 2.06 (d, J=8.53 Hz, 1 H) 2.10 – 2.21 (m, 1 H) 2.34 (s, 3 H) 2.40 – 2.48 (m, 1 H) 2.53 – 2.60 (m, 1 H) 2.61 – 2.70 (m, 2 H) 2.95 -3.01 (m, 1 H) 3.01 (s, 2 H) 3.10 – 3.16 (m, 1 H) 3.18 – 3.28 (m, 2 H) 3.72 (s, 1 H) 4.35 – 4.41 (m, 1 H) 4.46 – 4.70 (m, 2 H) 6.72 – 6.80 (m, 2 H) 7.05 – 7.23 (m, 6 H). Data for P-4 (R)-3-((3S,4S)-3-fiuoro-4-(4-hydroxyphenyl)piperidin-l-yl)-l-(4-methylbenzyl)pyrrolidin-2-one: LCMS (Method F) RT 2.11 min, m/z 383.2 (M+H+);; HPLC (Method C) RT 6.50 min, (Method D) RT 8.21 min; chiral HPLC (method C-6) RT 6.31 min; 1H NMR (400 MHz, methanol-d4) δ ppm 1.81 (dd, J=7.28, 2.76 Hz, 2 H) 2.06 (d, J=9.04 Hz, 2 H) 2.33 (s, 3 H) 2.43 (s, 1 H) 2.55 (br s, 1 H) 2.66 (d, J=40.16 Hz, 2 H) 2.75 – 2.80 (m, 1 H) 2.96 – 3.10 (m, 2 H) 3.20 – 3.28 (m, 2 H) 3.41 (d, J=5.52 Hz, 1 H) 3.66 – 3.75 (m, 1 H) 4.31 – 4.41 (m, 1 H) 4.46 – 4.71 (m, 2 H) 6.76 (d, J=8.53 Hz, 2 H) 7.05 – 7.23 (m, 6 H).
PATENT
Example 46 (Peak-1, Peak-2, Peak-3, Peak-4)
(S)-3-((3R,4R)-3-Fluoro-4-(4-hydroxyphenyl)piperidin-1-yl)-1-(4-methylbenzyl)pyrrolidin-2-one and (R)-3-((3R,4R)-3-fluoro-4-(4-hydroxyphenyl)piperidin-1-yl)-1-(4-methylbenzyl)pyrrolidin-2-one

(S)-3-((3S,4S)-3-Fluoro-4-(4-hydroxyphenyl)piperidin-1-yl)-1-(4-methylbenzyl)pyrrolidin-2-one and (R)-3-((3S,4S)-3-fluoro-4-(4-hydroxyphenyl)piperidin-1-yl)-1-(4-methylbenzyl)pyrrolidin-2-one

Step A. (±)-rel-(3S,4S)-1-benzyl-4-(4-methoxyphenyl)piperidin-3-ol

Step B. (±)-rel-(3S,4S)-4-(4-methoxyphenyl)piperidin-3-ol

Step C. (±)-rel-(3S,4S)-tert-butyl 4-(4-(tert-butoxycarbonyloxy)phenyl)-3-hydroxypiperidine-1-carboxylate

Step D. (±)-rel-(3S,4S)-tert-butyl 3-hydroxy-4-(4-hydroxyphenyl)piperidine-1-carboxylate

Step E. (3S,4S)-tert-Butyl 3-hydroxy-4-(4-hydroxyphenyl)piperidine-1-carboxylate and (3R,4R)-tert-butyl 3-hydroxy-4-(4-hydroxyphenyl)piperidine-1-carboxylate

Step F. (3R,4R)-tert-Butyl 4-(4-(benzyloxy)phenyl)-3-hydroxypiperidine-1-carboxylate

Step G. (3R,4R)-4-(4-(Benzyloxy)phenyl)piperidin-3-ol hydrochloride

Step H. 3-((3R,4R)-4-(4-(Benzyloxy)phenyl)-3-hydroxypiperidin-1-yl)-1-(4-methylbenzyl)pyrrolidin-2-one

Step I. 3-((3R,4R)-4-(4-(Benzyloxy)phenyl)-3-fluoropiperidin-1l-yl)-1-(4-methylbenzyl)pyrrolidin-2-one

Step J. 3-((3R,4R)-3-Fluoro-4-(4-hydroxyphenyl)piperidin-1-yl)-1-(4-methylbenzyl)pyrrolidin-2-one

Step K. (S)-3-((3R,4R)-3-Fluoro-4-(4-hydroxyphenyl)piperidin-1-yl)-1-(4-methylbenzyl)pyrrolidin-2-one and (R)-3-((3R,4R)-3-fluoro-4-(4-hydroxyphenyl)piperidin-1-yl)-1-(4-methylbenzyl)pyrrolidin-2-one
Step L. (3S,4S)-tert-Butyl 3-fluoro-4-(4-hydroxyphenyl)piperidine-1-carboxylate

Step M. 4-((3S,4S)-3-Fluoropiperidin-4-yl)phenol hydrochloride

Step N. 3-((3S,4S)-3-Fluoro-4-(4-hydroxyphenyl)piperidin-1-yl)-1-(4-methylbenzyl)pyrrolidin-2-one

Step O. (S)-3-((3S,4S)-3-Fluoro-4-(4-hydroxyphenyl)piperidin-1-yl)-1-(4-methylbenzyl)pyrrolidin-2-one and (R)-3-((3S,4S)-3-fluoro-4-(4-hydroxyphenyl)piperidin-1-yl)-1-(4-methylbenzyl)pyrrolidin-2-one
ADDITIONAL INFORMATION
Intravenous administration of BMS-986169 or BMS-986163 dose-dependently increased GluN2B receptor occupancy and inhibited in vivo [3H](+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine ([3H]MK-801) binding, confirming target engagement and effective cleavage of the prodrug. BMS-986169 reduced immobility in the mouse forced swim test, an effect similar to intravenous ketamine treatment. Decreased novelty suppressed feeding latency, and increased ex vivo hippocampal long-term potentiation was also seen 24 hours after acute BMS-986163 or BMS-986169 administration. BMS-986169 did not produce ketamine-like hyperlocomotion or abnormal behaviors in mice or cynomolgus monkeys but did produce a transient working memory impairment in monkeys that was closely related to plasma exposure. Finally, BMS-986163 produced robust changes in the quantitative electroencephalogram power band distribution, a translational measure that can be used to assess pharmacodynamic activity in healthy humans. Due to the poor aqueous solubility of BMS-986169, BMS-986163 was selected as the lead GluN2B NAM candidate for further evaluation as a novel intravenous agent for TRD.
REFERENCES
1: Bristow LJ, Gulia J, Weed MR, Srikumar BN, Li YW, Graef JD, Naidu PS, Sanmathi
C, Aher J, Bastia T, Paschapur M, Kalidindi N, Kumar KV, Molski T, Pieschl R,
Fernandes A, Brown JM, Sivarao DV, Newberry K, Bookbinder M, Polino J, Keavy D,
Newton A, Shields E, Simmermacher J, Kempson J, Li J, Zhang H, Mathur A, Kallem
RR, Sinha M, Ramarao M, Vikramadithyan RK, Thangathirupathy S, Warrier J, Islam
I, Bronson JJ, Olson RE, Macor JE, Albright CF, King D, Thompson LA, Marcin LR,
Sinz M. Preclinical Characterization of
(R)-3-((3S,4S)-3-fluoro-4-(4-hydroxyphenyl)piperidin-1-yl)-1-(4-methylbenzyl)pyrr
olidin-2-one (BMS-986169), a Novel, Intravenous, Glutamate N-Methyl-d-Aspartate
2B Receptor Negative Allosteric Modulator with Potential in Major Depressive
Disorder. J Pharmacol Exp Ther. 2017 Dec;363(3):377-393. doi:
10.1124/jpet.117.242784. Epub 2017 Sep 27. PubMed PMID: 28954811.
2. BMS-986163, a Negative Allosteric Modulator of GluN2B with Potential Utility in Major Depressive Disorder
Lawrence R. Marcin, Jayakumar Warrier, Srinivasan Thangathirupathy, Jianliang Shi, George N. Karageorge, Bradley C. Pearce, Alicia Ng, Hyunsoo Park, James Kempson, Jianqing Li, Huiping Zhang, Arvind Mathur, Aliphedi B. Reddy, G. Nagaraju, Gopikishan Tonukunuru, Grandhi V. R. K. M. Gupta, Manjunatha Kamble, Raju Mannoori, Srinivas Cheruku, Srinivas Jogi, Jyoti Gulia, Tanmaya Bastia, Charulatha Sanmathi, Jayant Aher, Rajareddy Kallem, Bettadapura N. Srikumar, Kumar Kuchibhotla Vijaya, Pattipati S. Naidu, Mahesh Paschapur, Narasimharaju Kalidindi, Reeba Vikramadithyan, Manjunath Ramarao, Rex Denton, Thaddeus Molski, Eric Shields, Murali Subramanian, Xiaoliang Zhuo, Michelle Nophsker, Jean Simmermacher, Michael Sinz, Charlie Albright, Linda J. Bristow, Imadul Islam, Joanne J. Bronson, Richard E. Olson, Dalton King, Lorin A. Thompson, and John E. Macor
Publication Date (Web): April 13, 2018 (Letter)
DOI: 10.1021/acsmedchemlett.8b00080
| Patent ID | Patent Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9221796 | Selective NR2B antagonists |
2015-01-05
|
2015-12-29
|
//////////////////BMS-986169, BMS-986169, BMS 986169, BMS986169, preclinical
O=C1N(CC2=CC=C(C)C=C2)CC[C@H]1N3C[C@@H](F)[C@H](C4=CC=C(O)C=C4)CC3