















Centanafadine; UNII-D2A6T4UH9C; EB-1020 free base; D2A6T4UH9C; 924012-43-1
CTN SR; EB-1020; EB-1020 SR
WO 2007016155
| Molecular Formula: | C15H15N |
|---|---|
| Molecular Weight: | 209.292 g/mol |
- Phase II Attention-deficit hyperactivity disorder
- No development reported Major depressive disorder; Neuropathic pain
Most Recent Events
- 20 Dec 2016 Neurovance plans a phase III trial for Attention-deficit hyperactivity disorder
- 27 Jul 2016 Efficacy data from a phase IIb trial in Attention-deficit hyperactivity disorder released by Neurovance
- 16 Jul 2016 No recent reports of development identified for phase-I development in Attention-deficit-hyperactivity-disorder in Canada (PO)
- Originator Euthymics Bioscience
- Developer Euthymics Bioscience; Neurovance
- Class Azabicyclo compounds; Cyclohexanes; Naphthalenes; Small molecules
- Mechanism of Action Adrenergic uptake inhibitors; Dopamine uptake inhibitors; Serotonin uptake inhibitors


cas 923981-14-0 hydrochloride
| Molecular Formula: | C15H16ClN |
|---|---|
| Molecular Weight: | 245.75 g/mol |
(1R,5S)-1-(naphthalen-2-yl)-3-azabicyclo(3.1.0)hexane hydrochloride
Centanafadine (INN) (former developmental code name EB-1020) is a serotonin-norepinephrine-dopamine reuptake inhibitor (SNDRI) under development by Neurovance in collaboration with Euthymics Bioscience as a treatment for attention deficit hyperactivity disorder (ADHD) that inhibits the reuptake of norepinephrine, dopamine and serotonin with a ratio of 1:6:14, respectively.[1][2][3] As of August 2015, it is in phase II clinical trials.[1]
Also claimed is their use for treating attention deficit hyperactivity disorder (ADHD), fragile-X associated disorder, autism spectrum disorder and depression. See WO2015089111, claiming method for treating fragile X-associated disorders, assigned to Neurovance, naming Piskorski, Bymaster and Mckinney. Neurovance, an affiliate of Euthymics Bioscience, is developing centanafadine, a sustained release formulation and a non-stimulant triple reuptake inhibitor, for treating ADHD and is also investigating the drug for treating neuropathic pain.
In June 2015, the drug was reported to be in phase 2 clinical and preclinical development for treating ADHD and neuropathic pain, respectively. Inventors are affiliated with Neurovance.
Attention-deficit hyperactivity disorder (ADHD) is a central nervous system
(CNS) disorder characterized by developmentally inappropriate inattention, hyperactivity, and impulsivity (Buitelaar et al., 2010; Spencer et al., 2007). ADHD is one of the most common developmental disorders in children with 5-10% prevalence (Scahill et al., 2000; Polanczyk et al., 2007). While ADHD was once regarded as only a childhood disorder, it can continue through adolescence and into adulthood. An estimated 2.9-4.4% of the adult population has continuing ADHD (Kessler et al., 2006; Faraone and Biederman, 2005). Major symptoms in adults include inattention, disorganization, lack of concentration and to some extent impulsivity, which result in difficulty functioning, low educational attainment, under achievement in vocational and educational pursuits, and poor social and family relations (Biederman et al, 2006; Barkely et al., 2006).
The exact causes of ADHD are not known, but a dysfunction of the prefrontal cortex and its associated circuitries has been posited as a key deficit in ADHD (Arnsten, 2009). Consistent with this notion is the finding that abnormal catecholaminergic function plays a key role, particularly in prefrontal cortical regions (Arnsten 2009). The
catecholamines norepinephrine (NE) and dopamine (DA) are highly involved in several domains of cognition including working memory, attention, and executive function. Accordingly, these monoamine neurotransmitters are believed to work in concert in modulating cognitive processes.
Pharmacotherapy is a primary form of treatment utilized to reduce the symptoms of ADHD. Stimulants such as methylphenidate and amphetamines are commonly used for ADHD. The major mechanism of action of the stimulants is inhibition of DA and NE transporters. The stimulants are effective against the core symptoms of ADHD and have a response rate of about 70% (Spencer et al. , 2005). However, major concerns about stimulants include risk of abuse, dependency, and diversion as well as potential neurotoxic effects of amphetamines (Berman et al., 2009). The abuse potential of stimulants is particularly problematic in adults because substance abuse is a common co-morbidity with adult ADHD (Levin and Kleber, 1995; Ohlmeier, 2008).
Another major drug used to treat ADHD is atomoxetine, which is a selective norepinephrine reuptake inhibitor. Major advantages of atomoxetine compared to the stimulants is lack of abuse potential, once-daily dosage, and superior treatment of comorbidities such as anxiety and depression. However, atomoxetine has lower efficacy and takes 2-4 weeks for onset of action (Spencer et al., 1998; Newcorn et al., 2008).
Accordingly, there remains a need for effective pharmaceuticals which may be used in the treatment of ADHD and other conditions affected by monoamine neurotransmitters.
PATENT
WO 2007016155
https://www.google.ch/patents/WO2007016155A2?hl=de&cl=en
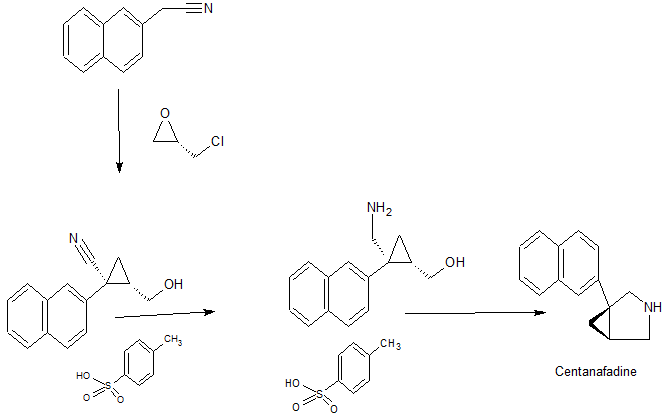
Reaction Scheme 1 below generally sets forth an exemplary process for preparing l-aryl-3-azabicyclo[3.1.0] hexane analogs from the corresponding 2-bromo-2- arylacetate or 2-chloro-2-arylacetate. The bromo or chloro acetate react with acrylonitrile to provide the methyl 2-cyano-l-arylcyclopropanecarboxylate, which is then reduced to the amino alcohol by reducing agents such as lithium aluminum hydride (LAH) or sodium aluminum hydride (SAH) or NaBH4 with ZnCl2. Cyclization of the amino alcohol with SOCl2 or POCl3 will provide the l-aryl-3-azabicyclo[3.1.0]hexane. The cyclization of substituted 4-aminobutan-l -ol by SOCl2 or POCl3 into the pyrrolidine ring system was reported by Armarego et al, J. Chem. Soc. [Section C: Organic] 19:3222-9, (1971), and in Szalecki et al., patent publication PL 120095 B2, CAN 99:158251. Oxalyl chloride, phosphorous tribromide, triphenylphosphorous dibromide and oxalyl bromide may be used for the same purpose. The methyl 2-bromo-2 -arylacetate or methyl 2- chloro-2-arylacetate may be synthesized from subsituted benzoylaldehyde or methyl-2- arylacetate as shown in Reaction Scheme IA.
Reaction Scheme 1

Reduction

Reagents: (a) NaOMe; (b) LiAIH4; (c) SOCI2; (d) POCI3; (e) NaOH or NH3 H2O
Reaction Scheme IA


Reagents: (a) CHCI3, NaOH; (b) SOCI2; (c) MeOH; (d) NaBrO3, NaHSO3 [00138] Reaction Scheme 2 below illustrates another exemplary process for transforming methyl 2-cyano-l-arylcyclopropanecarboxylate to a desired compound or intermediate of the invention. Hydrolysis of the cyano ester provides the potassium salt which can then be converted into the cyano acid. Reduction and cyclization of the 2- cyano-1-arylcyclopropanecarboxylic acid with LAH or LiAlH(OMe)3according to the procedure outlined in Tetrahedron 45:3683 (1989), will generate l-aryl-3- azabicyclo[3.1.0]hexane. In addition, the cyano- 1-arylcyclopropanecarboxylic acid can be hydrogenated and cyclized into an amide, which is then reduced to l-aryl-3- azabicyclo[3.1.0]hexane.
Reaction Scheme 2

Hydrolysis

Reagents: (a) NaOMe; (b) KOH; (c) HCI; (d) LiAIH(OMe)3, or LAH, or SAH, then HCI; (e) H2/Pd or H2/Ni
[00139] Reaction Scheme 3 below discloses an alternative exemplary process for converting the methyl 2-cyano-l-arylcyclopropanecarboxylate to a desired compound or intermediate of the invention. The methyl 2-cyano-l-arylcyclopropanecarboxylate is reduced and cyclized into l-aryl-3-aza-bicyclo[3.1.0]hexan-2~one, which is then reduced to l-aryl-3-azabicyclo[3.1.0]hexane [Marazzo, A. et al., Arkivoc 5:156-169, (2004)].
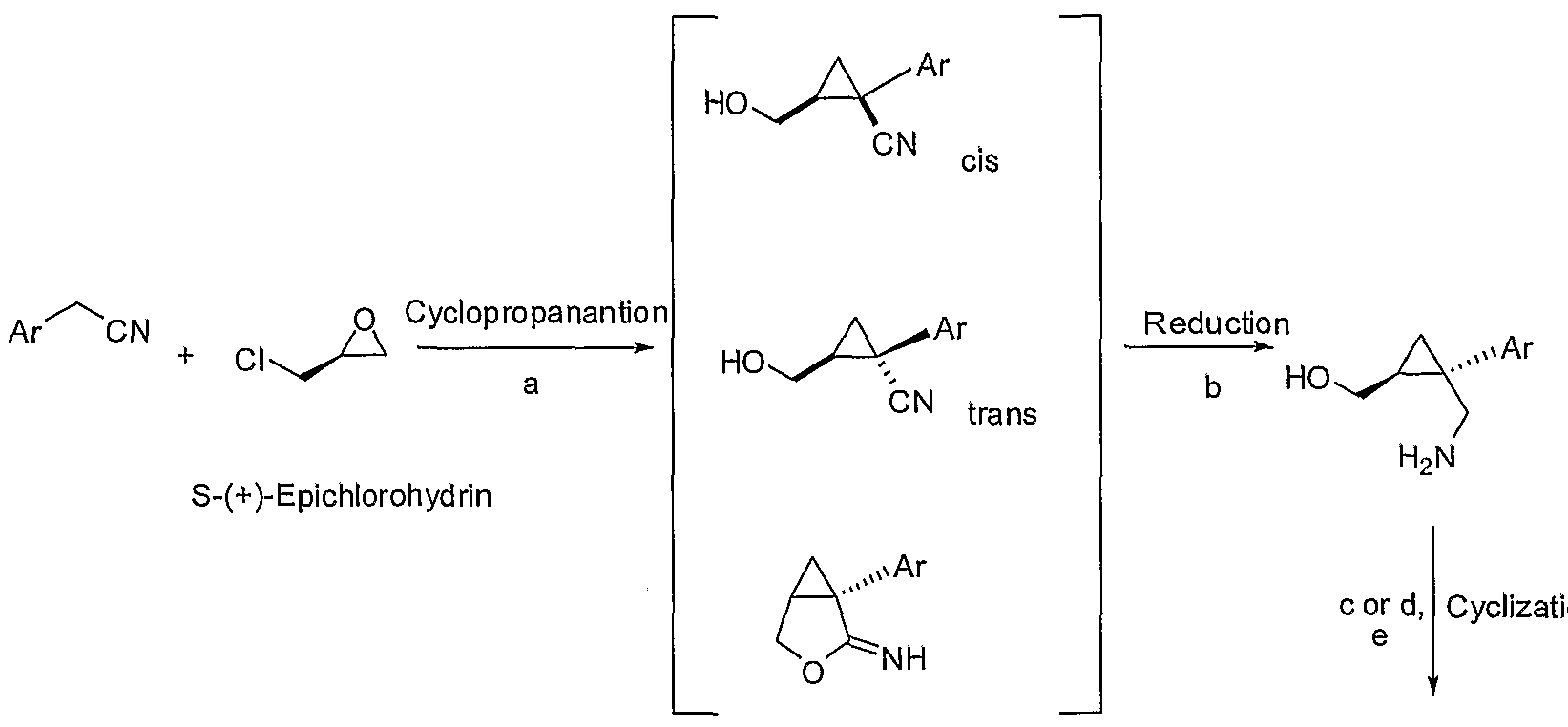
Reaction Scheme 3

Reagents: (a) H2/Pd or H2/Ni; (b) B2H6 or BH3 or LAH, then HCI [00140] Reaction Scheme 4 below provides another exemplary process to prepare l-aryl-3-azabicyclo[3.1.0] hexane analogs. Reaction of 2-arylacetonitrile with (+)- epichlorohydrin gives approximately a 65% yield of 2-(hydroxyrnethyl)-l- arylcyclopropanecarbonitrile (85% cis) with the trans isomer as one of the by-products [Cabadio et al., Fr. Bollettino Chimico Farmaceutico 117:331-42 (1978); Mouzin et al., Synthesis 4:304-305 (1978)]. The methyl 2-cyano-l-arylcyclopropanecarboxylate can then be reduced into the amino alcohol by a reducing agent such as LAH, SAH or NaBH4 with ZnCl2 or by catalytic hydrogenation. Cyclization of the amino alcohol with SOCl2 or POCl3 provides the l-aryl-3-azabicyclo[3.1.0]hexane. The cyclization of substituted 4-aminobutan-l-ol by SOCl2 or POCl3 into the pyrrolidine ring system has been reported previously [Armarego et al., J. Chem. Soc. [Section C: Organic] 19:3222-9 (1971); patent publication PL 120095 B2, CAN 99:158251).
ϋv siυjiijJsoLJa
Reaction Scheme 4
Ar CN
ion

Reagents: (a) NaHMDS; (b) LAH or catalytic hydrogenation; (c) SOCl2; (d) POCI3; (e) NaOH

[00141] Reaction Scheme 5 provides an exemplary process for synthesizing the
(IR, 5S)-(+)-l-aryl-3-azabicyclo[3.1.0]hexanes. Using (S)-(+)-epichlorohydrin as a starting material in the same process described in Scheme 4 will ensure a final product with 1-R chirality [Cabadio, S. et al, Fr. Bollettino Chimico Farmaceutico 117:331-42 (1978)].
Reaction Scheme 5
ion
^Ar
H’..
Reagents: (a) NaHMDS; (b) LAH or catalytic hydroge nation; (c) SOCI2; (d) POCl3; (e) NaOH j_j
[00142] Reaction Scheme 6 provides an exemplary process to prepare the (1 S,5R)-
(-)-l-aryl-3-azabicyclo[3.1.0]hexanes. Using (R)-(-)-epichlorohydrin as a starting material in the same process described in Scheme 4 will ensure a final product with 1-S chirality [Cabadio, S. et al, Fr. Bollettino Chimico Farmaceutico 117:331-42 (1978)].
Reaction Scheme 6
Ar CN

c or d, Cyclization

Reagents: (a) NaHMDS; (b) LAH or catalytic hydrogenation; (C) SOCI2; (d) POCI3; (e) NaOH

[00143] Reaction Scheme 7 provides an alternative exemplary process for transforming the 2-(hydroxymethyl)-l-arylcyclopropanecarbonitrile to a desired compound or intermediate of the invention via an oxidation and cyclization reaction. Utilizing chiral starting materials (+)-epichlorohydrin or (-)-epichlorohydrin will lead to the corresponding (+)- or (-)-enantiomers and corresponding chiral analogs through the same reaction sequences.
Reaction Scheme 7
O Cyclopropanantion Oxidation
Ar CN
CK Ar Ar a HO HO
CN
65% yield, 88% cis O
Hydrogenation
C Cyclization

Reagents: (a) NaNH2; (b) KMnO4; (c) H2/Ni or Pt; (d) B2H6 Or BH3 Or LAH, then HCI

[00144] Reaction Scheme 8 provides an exemplary process for transforming the epichlorohydrin to a desired compound or intermediate of the invention via a replacement and cyclization reaction. The reaction of methyl 2-arylacetate with epichlorohydrin gives methyl 2-(hydroxymethyl)~l~arylcyclopropanecarboxylate with the desired cis isomer as the major product. The alcohol is converted into an OR3 group such as -O-mesylate, -O- tosylate, -O-nosylate, -O-brosylate, -O-trifluoromethanesulfonate. Then OR3 is replaced by a primary amine NH2R4, where R4 is a nitrogen protection group such as a 3,4- dimethoxy-benzyl group or other known protection group. Nitrogen protecting groups are well known to those skilled in the art, see for example, “Nitrogen Protecting Groups in Organic Synthesis”, John Wiley and Sons, New York, N.Y., 1981, Chapter 7; “Nitrogen Protecting Groups in Organic Chemistry”, Plenum Press, New York, N.Y., 1973, Chapter 2; T. W. Green and P. G. M. Wuts in “Protective Groups in Organic Chemistry”, 3rd edition, John Wiley & Sons, Inc. New York, N. Y., 1999. When the nitrogen protecting group is no longer needed, it may be removed by methods well known in the art. This replacement reaction is followed by a cyclization reaction which provides the amide, which is then reduced into an amine by a reducing agent such as LAH. Finally the protection group is removed to yield the l-aryl-3- azabicyclo[3.1.0]hexane analogs. Utilizing chiral (S)-(+)-epichlorohydrin as a starting material leads to the (lR,5S)-(+)-l-aryl-3-azabicyclo[3.1.0]hexane analogs with the same reaction sequence. Similarly, the (R)-(-)-epichlorohydrin will lead to the (lS,5R)-(-)-l- aryl-3-azabicyclo[3.1.0]hexane analogs.
Reaction Scheme 8
O Cyclop ro pa nantion
Ar CO2Me + C|v Ar Ar
HO R3O
CO2Me CO2Me
Replacement Cyclization

Reagents: (a) NaNH2; (b) MsCI; (c) R4NH2; (d) LAH or SAH or BH3; (e) HCI

[00145] Reaction Scheme 9 provides an exemplary process for transforming the diol to a desired compound or intermediate of the invention. Reduction of the diester provides the diol which is then converted into an OR3 group such as -O-mesylate, -O- tosylate, -O-nosylate, -O-brosylate, -O-trifluoromethanesulfonate. Then OR3 is replaced by a primary amine NH2R6, where R6 is a nitrogen protection group such as a 3,4- dimethoxy-benzyl group or other protection groups known in the art (e.g., allyl amine, tert-butyl amine). When the nitrogen protecting group is no longer needed, it may be removed by methods known to those skilled in the art.
Reaction Scheme 9

X=CI or Br

Deprotection ft* Replacement Cyclization
Reagents: (a) NaOMe; (b) NaBH4; (c)MsCI; (d) NH3, then HCI; (e) R6NH2; (f) H2/Pd or acid deprotection, then HCI
[00146] Reaction Scheme 10 provides an exemplary process for resolving the racemic l-aryl-3-aza-bicyclo[3.1.0]hexane to enantiomers. The resolution of amines through tartaric salts is generally known to those skilled in the art. For example, using O,O-Dibenzoyl-2R,3R-Tartaric Acid (made by acylating L(+)-tartaric acid with benzoyl chloride) in dichloroethane/methanol/water, racemic methamphetamine can be resolved in 80-95% yield, with an optical purity of 85-98% [Synthetic Communications 29:4315- 4319 (1999)]. Reaction Scheme 10

Racemate (1 R, 5S)-enantiomer

Racemate (1 S, 5R)-enantiomer
Reagents: (a) L-(-)-DBTA; (b) NaOH, then HCI in IPA; (c) D-(+)-DBTA
[00147] Reaction Scheme 11 provides an exemplary process for the preparation of
3-alkyl-l-aryl-3-azabicyclo[3.1.0]hexane analogs. These alkylation or reductive animation reaction reagents and conditons are generally well known to those skilled in the art.
Reaction Scheme 11

R= Me, Et, Propyl, i-propyl, cyclopropyl, i-butyl, etc.
[00148] Enantiomers of compounds within the present invention can be prepared as shown in Reaction Scheme 12 by separation through a chiral chromatography. Reaction Scheme 12

[00149] Alternatively, enantiomers of the compounds of the present invention can be prepared as shown in Reaction Scheme 13 using alkylation reaction conditions exemplified in scheme 11.
Reaction Scheme 13


[00150] Reaction Scheme 14 provides an exemplary process for preparing some N- methyl l-aryl-3-aza-bicyclo[3.1.0]hexane analogs. The common intermediate N-methyl bromomaleide is synthesized in one batch followed by Suzuki couplings with the various substituted aryl boronic acids. Cyclopropanations are then carried out to produce the imides, which are then reduced by borane to provide the desired compounds.
Reaction Scheme 14


Reagents and conditions: (a) MeNH2, THF, 10 0C, 1.5 hr; (b) NaOAc, Ac2O1 60 0C, 2 hr; (c) PdCI2C dppf), CsF, dioxane, 40 0C, 1-6 hr; (d) Me3SOCI, NaH, THF, 50-65 0C, 2-6 hr; (e) 1M BH3/THF, O 0C; 60 0C 2 hr (f) HCI, Et2O
[00151] Reaction Scheme 15 provides an additional methodology for producing 1- aryl-3-azabicyclo[3.1.0] hexanes.
Reaction Scheme 15


[00152] Reaction Scheme 16 provides an additional methodology for producing 1- aryl-3-azabicyclo[3.1.0] hexanes. Reaction scheme 16


[00153] Reaction Scheme 17 provides an additional methodology for producing 1- aryl-3-azabicyclo[3.1.0] hexanes.
Reaction Scheme 17

[00154] Reaction Scheme 18 provides an additional methodology for producing 1- aryl-3-azabicyclo[3.1.0] hexanes. Utilizing chiral starting materials (+)-epichlorohydrin or (-)-epichlorohydrin will lead to the corresponding chiral analogs through the same reaction sequences. Reaction Scheme 18


[00155] Reaction Scheme 19 provides an additional methodology for producing 1- aryl-3-azabicyclo[3.1.0] hexanes.
Reaction Scheme 19

R= propyl , butyl, etc.
[00156] Reaction Scheme 20 provides an additional methodology for producing 1- aryl-3-azabicyclo[3.1.0] hexanes.
Reaction Scheme 20
H

Ac2O NaOAc, reflux

R= ferf-butyl, etc.
[00157] Reaction Scheme 21 provides an additional methodology for producing 3- and/or 4-subsitituted l-aryl-3-azabicyclo[3.1.0] hexanes. Reaction Scheme 21

(BoC)2O DCM

R= methyl, etc. -Ar v Ar R1 = methyl, etc. R- N H HCI
R • HCI
[00158] Reaction Scheme 22 provides an additional methodology for producing 3- and/or 4-subsitituted l-aryl-3-azabicyclo[3.1.0] hexanes.
Reaction Scheme 22

(BoC)2O DCM
1. 2.

R= Ri

[00159] Reaction Scheme 23 provides an additional methodology for producing 3- and/or 2-subsitituted 1 -aryl-3-azabicyclo[3.1.0] hexanes.
Reaction Scheme 23

KBH4
R = Me, etc. MeOH R1 = Me, etc.

HCI HCI Ether Ether

[00160] Reaction Scheme 24 provides an additional methodology for producing 2- and/or 3 -substituted l-aryl-3-azabicyclo[3.1.0] hexanes.
Reaction Scheme 24
I) TMSCI; PhMe
Et3N; NaBH3CN 2) R2Li EtOH





[00161] Reaction Scheme 25 provides an additional generic methodology for producing 1 -aryl-3 -azabicyclo[3.1.0] hexanes .
Reaction Scheme 25
Ar
Cyolopropanation Ar Reduction Ar Cyciization / \
Ar CN + Cl HO. HO
CN
H2N
or Protection

[00162] Reaction Scheme 26 provides another generic methodology for producing l-aryl-3-azabicyclo[3.1.0] hexanes.
Reaction Scheme 26
0

Reduction Deprotection/ dealkylation


C. Synthesis of various naphthyl and phenyl 3-azabicyclo[3.1.01hexane Hydrochlorides
(1) Synthesis of lS,5R-(-Vl-(l-naphthylV3-azabicyclol3.1.01hexane Hydrochloride as Representative Procedure for (l)-(6).

[00340] To a stirring solution of ( 1 R,2S)-(2-Aminomethyl-2-( 1 – naphthyl)cyclopropyl)-methanol prepared according to Example XIVB(I) above (3.2 g, 0.014 moles) in 35 niL of dichloroethane (DCE), at room temperature under nitrogen, was added 1.2 niL (0.017 moles, 1.2 eq) of SOCl2 slowly via syringe while keeping the temperature below 50 0C. (Note: The reaction exotherms from 22 0C to 45 0C) The resulting mixture was stirred for 3.5 h at room temperature after which time, TLC analysis (SiO2 plate, CH2Cl2/MeOH/NH4OH (10:1:0.1)) showed no starting material remaining. The mixture was quenched with 40 mL of water and the layers were separated. The organic layer was washed with H2O (2 x 5O mL). The aqueous layers were combined, made basic with ION NaOH to pH = 10 (pH paper) and extracted with 2 x 100 mL of CH2Cl2. The combined organics were dried over Na2SO4, filtered and concentrated to an oil. The oil was dissolved in MeOH (20 mL), treated with 15 mL of 2M HCl/Et2O and concentrated in vacuo to a suspension. The slurry was diluted with 25 mL of Et2O, filtered and washed with 35 mL of Et2O. The solid product was dried overnight (-29 mmHg, 5O0C) to give 1 g (29%) of pure product as a white solid. 1H NMR (400 MHz, CDCl3) δ 1.22 (t, J=7.37 Hz, 1 H), 1.58 (dd, J=6.00, 4.73 Hz, 1 H), 2.03 – 2.10 (m, 1 H), 3.25 – 3.27 (m, 1 H), 3.42 (d, J=I 1.52 Hz, 1 H), 3.64 (d, J=I 1.62 Hz, 1 H), 3.74 – 3.85 (m, 2 H), 7.32 – 7.39 (m, 1 H), 7.40 – 7.48 (m, 2 H), 7.48 – 7.55 (m, 1 H), 7.75 (d, J=8.20 Hz, 1 H), 7.79 – 7.85 (m, 1 H), 8.04 (d, J=8.30 Hz, 1 H), 13C NMR (101 MHz, CDCl3) δ 14.54, 22.43, 30.89, 48.01, 51.89, 123.92, 125.60, 126.24, 126.93, 129.04, 129.17, 133.55, 134.04, LC/MS (m/z M+1) 210.0, [α]D (c=l, MeOH), = -54.4.
(2) lR,5S-(+)-l-g-naphthyl)-3-azabicvclof3,1.01hexane Hydrochloride

[00341] Yield = 29%; 1H NMR (400 MHz, METHANOL-^) δ 1.24 – 1.32 (m, 1
H), 1.32 – 1.37 (m, 1 H), 2.23 – 2.31 (m, 1 H), 3.47 (d, J=11.71 Hz, 1 H), 3.66 (d, J=11.71 Hz, 1 H), 3.85 (d, J=11.62 Hz, 1 H), 3.93 (dd, J=11.67, 3.95 Hz, 1 H), 7.46 (dd, J=8.25, 7.08 Hz, 1 H), 7.50 – 7.57 (m, 1 H), 7.57 – 7.65 (m, 2 H), 7.86 (d, J=8.30 Hz, 1 H), 7.89 – 7.95 (m, 1 H), 8.17 (d, J=8.49 Hz, 1 H), 13C NMR (101 MHz, METHANOL-^) δ 22.36, 30.65, 30.65, 48.09, 51.99, 123.78, 125.47, 125.89, 126.50, 128.65, 128.88, 133.87, 134.28, LC/MS (m/z M+1 210.0), [α]D (c=l, MeOH), = + 55.6.
(4) lR.5S-(+)-l-(2-naphthylV3-azabicvclo[3.1.01hexane Hydrochloride

[00343] Yield = 30%; 1H NMR (400 MHz, DMSO-J6) δ 1.14 – 1.23 (m, 1 H), 1.44
– 1.50 (m, 1 H), 2.17 – 2.26 (m, 1 H), 3.36 – 3.43 (m, 1 H), 3.47 – 3.61 (m, 2 H), 3.75 (d, J-11.23 Hz, 1 H), 7.36 (dd, J=8.59, 1.85 Hz, 1 H), 7.42 – 7.53 (m, 2 H), 7.80 (d, J=1.56 Hz, 1 H), 7.82 – 7.90 (m, 3 H), 9.76 (br. s., 1 H), 13C NMR (101 MHz, DMSO-J6) δ 16.41, 24.11, 31.36, 47.50, 49.97, 125.43, 125.76, 126.41, 127.04, 128.07, 128.15, 128.74, 132.39, 133.55, 137.62, ), LC/MS (m/z M+1 210.1 , [α]D (c=l, MeOH), = + 66.0.
PATENT
WO 2008013856
https://www.google.com/patents/WO2008013856A2?cl=en
The compound (-^-(S^-dichlorophenylJ-S-azabicyclotS.l.Olhexane and its pharmaceutically acceptable salts have been previously described as agents for treating or preventing a disorder alleviated by inhibiting dopamine reuptake, such as depression (See, US Patent Nos. 6,569,887 and 6,716,868). However, available methods for synthesizing (-)-l-(334-dichlorophenyl)-3-azabicyclo[3.1.0]hexanes and other l-aryl-3-azabicyclo[3.1.0]hexanes are presently limited.
US Patent No. 4,231,935 (Example 37) describes the synthesis of racemic (±)- l-(3,4-dichlorophenyl)-3-azabicyclo[3.1.0]hexane hydrochloride according to the following scheme.


US Patent Nos. 6,569,887 and 6,716,868 describe the preparation of (-)-l-(3,4- dichlorophenyl)-3-azabicyclo[3.1.0]hexane by resolution of racemic (±)-l-(3,4- dichlorophenyl)-3-azabicyclo[3.1.0]hexane hydrochloride using a chiral polysaccharide stationary phase. The foregoing methods provide limited tools for producing (-)-l-(3,4- dichlorophenyl)-3-azabicyclo[3.1.0] hexane and other 1-aryl— 3- azabicyclo[3.1.0]hexanes, underscoring a need for additional methods and compositions to produce the compounds.
Example VI Preparation of ClR. 5S)-l-naphthalen-2-yl-3-azabicyclof3.1.0“|hexane hydrochloride
using Reaction Schemes 1 & 12
A. Synthesis of (IR. 2SV2-Hvdroxymethyl-2-naphthyl- cvclopropancarbonitrile

To a stirring solution of 2-naphthylacetonitrile (50.0 g, 0.299 moles) and (S)-
(+)-epichlorohydrin (36.0 g, 0389 moles) in anhydrous THF (300 mL) at -15 to -20 0C under nitrogen, was added sodium bis (trimethylsilyl)amide (2M in THF, 300 mL, 0.600 moles) slowly via addition funnel while keeping the temperature between -15 0C and —20 °C. After completion of the addition, the mixture was stirred for 3 hours at -15 0C to -20 °C. The reaction mixture was quenched by slow addition of 2M HCl (520 mL) allowing the temperature to rise to 15 0C as the neutralization proceeded. The layers were allowed to settle and the layers were separated. The aqueous layer was extracted once with ethyl acetate (30OmL). The organic portion was washed with brine (4000 mL) dried over sodium sulfate, filtered and concentrated under reduced pressure to provide an orange oil which was used without further purification. B. Synthesis of ((1S, 2R)-2-AminomethvI-2-naphthylen-2-yl cycIopropyD- methanol

To a solution of nitrile in THF (300 mL) was slowly added borane dimethylsulfide (10 M, 60 mL, 0.60 moles) via addition funnel. The reaction temperature was maintained below 60 0C during the addition. After completion of the addition, the reaction was held at 60 0C until the starting nitrile was completely consumed (approximately 2.5 hours). The mixture was cooled below 15 0C and 2M HCl (200 mL) was slowly added maintaining a temperature below 20 0C. The reaction mixture was then heated to 500C for one hour. After the heating period, the reaction was cooled below 300C and isopropyl acetate (200 mL) and water (250 mL) were added. The phases were separated and the organic phase was discarded. Ammonium hydroxide (75 mL) was added and the mixture cooled to 25 0C with stirring. The aqueous phase was extracted with isopropyl acetate (2x 250 mL). The combined organic phases were washed with 5% dibasic sodium phosphate (200 mL) and saturated NaCl (200 mL), dried over sodium sulfate and concentrated. The viscous yellow oil was dissolved in isopropyl acetate (500 mL) and heated to 55 0C with stirring. p-Toluene sulfonic acid monohydrate(54.25 g, 0.285 mole) was added over 5 minutes. A white solid formed as the acid was added. The reaction mixture was slowly cooled to room temperature, filtered and washed with isopropyl acetate. Yielded – 53.7 g white solid 45% (tosylate salt)
C. Synthesis of (IR. 5SM-naphthaIen-2-vI-3-azabicvclo[3.1.01hexane hydrochloride

To a stirring slurry of ((lS,2R)-2-aminomethyl-2-naphthylen-2-yl cyclopropyl)-methanol tosylate (53.7g, 0.134 mole) in isopropyl acetate (350 mL), at room temperature under nitrogen, was added thionyl chloride (11.8 mL, 0.161 moles) slowly via addition funnel while keeping the temperature below 35 0C. The resulting mixture was stirred for 1 hour, after which time, no starting material remained. The mixture was neutralized with the slow addition of 5 N NaOH (160 mL) keeping the temperature below 30 0C. The phases were separated and the aqueous phase was extracted with isopropyl acetate (200 mL). The combined organic extracts were washed with saturated sodium chloride (150 mL), dried over sodium sulfate, filtered and concentrated to 300 mL. The hydrochloride was made directly from this solution by slowly adding HCl in 2-propanol (5-6N, 26 mL). The mixture was stirred for 15 minutes and filtered and washed with isopropyl acetate. The wet cake was slurried in 2-propanol (400 mL) and heated to reflux with stirring under nitrogen for 2 hours. The resulting slurry was allowed to cool and stir at room temperature overnight. The resulting slurry was filtered and washed with 2-propanol. The solid was dried in a vacuum oven at 400C. Yield – 21.1 g, 64.2% 1H NMR (400 MHz, DMSCW6) d ppm 1.23 (t) 1.40 (t) 2.21 – 2.28 (m) 3.40 – 3.47 (m) 3.50 – 3.66 (m) 3.74 – 3.82 (m) 7.39 (dd) 7.44 – 7.55 (m) 7.82 (s) 7.84 – 7.92 (m) 9.33 (br. s.) 9.69 (br. s.). LC/MS (m/z M+1 210)
PATENT
WO 2013019271
https://google.com/patents/WO2013019271A1?cl=en
Example I
Preparation of (lR,5S)-(+)-l-(naphthalen-2-yl)-3-azabicyclo[3.1.0]hexane
[00108] (lR,5S)-(+)-l-(naphthalen-2-yl)-3-azabicyclo[3.1.0]hexane may be prepared as follows:
Step 1: Synthesis of [(lS.2R)-2-(aminomethyl)-2-(2-naphthyl‘)cvclopropyl]methan-l-oK p- toluenesulfonic acid salt [00109] 500g (2.99 mol, 1.0 eq) of 2-naphthylacetonitrile was charged to a 12 L 3- neck round bottom flask equipped with overhead stirrer, addition funnel, thermocouple, nitrogen inlet, cooling bath and drying tube. 3.0 L of tetrahydrofuran was added and stirred at room temperature to dissolve all solids. 360 g (3.89 mol, 1.30 eq) (S)-(+)-epichlorohydrin was added and then the solution was cooled to an internal temperature of – 25 °C. 3.0 L of a 2 molar solution of sodium bis(trimethylsilyl)amide in tetrahydrofuran (6.00 mol, 2.0 eq) was added to the reaction mixture via addition funnel at a rate such that the internal temperature of the reaction mixture is maintained at less than -15 °C. After completion of the addition, the mixture was stirred at between -20 °C and -14 °C for 2 hours 15 minutes. Borane- dimethylsulfide complex (750 mL of a 10.0 molar solution, 7.5 mol, 2.5 eq) was then slowly added to the reaction mixture at a rate such that the internal temperature was maintained at less than -5 °C. Upon completion of the borane-dimethylsulfide addition the reaction mixture was heated to an internal temperature of 60 °C and stirred overnight at this temperature. Additional borane-dimethylsulfide complex (75 mL, 0.75 mol, 0.25 eq) was then added and the reaction mixture stirred at 60 °C for 1 hour 45 minutes. The reaction mixture was cooled to room temperature and then quenched by slow addition into pre-cooled (3 °C) 2 molar aqueous hydrochloric acid (5.76 L, 11.5 mol, 3.8 eq) at a rate such that the temperature of the quench solution was maintained at less than 22 °C. The two phase mixture was then heated at an internal temperature of 50 °C for 1 hour followed by cooling to RT. Isopropyl acetate (2.0 L) and water (2.5 L) were added, the mixture agitated, and then the layers were allowed to settle. The upper organic layer was discarded. Aqueous ammonia (750 mL) was added to the aqueous layer which was then extracted with isopropylacetate (2.5 L). The aqueous layer was extracted with isopropylacetate (2.5 L) a second time. The organic extracts were combined and then sequentially washed with a 5% solution of sodium dibasic phosphate in water (2.0 L) followed by saturated brine (2.0 L). The organic layer was then concentrated to a total volume of 5.0 L and then heated to 50 °C. para-Toluene sulfonic acid monohydrate (541 g, 2.84 mol) was then added in portions. During the addition white solids precipitated and a mild exotherm was observed. Upon completion of the addition the mixture was allowed to cool to RT and the solids collected by filtration. The filtercake was washed twice with isopropylacetate, 1.0 L each wash. The filtercake was then dried to a constant weight to give 664.3 g (55% yield) of the desired product as a white solid. Step 2: Synthesis of (5S.lRVl-(2-naphthylV3-azabicvclor3.1.01hexane HC1 salt
[00110] The amine-tosylate salt from step 1 (2037.9 g, 5.10 mol) was suspended in isopropylacetate ( 13.2 L) to give a white slurry in a 50 L 3 -neck RB equipped with an overhead stirrer, thermocouple, addition funnel, nitrogen inlet and drying tube.
Thionylchloride (445 mL, 6.12 mol, 1.20 eq) was then added via addition funnel over one hour 5 minutes. The maximum internal temperature was 24 °C. After stirring for 4 hours 15 minutes 5 molar aqueous sodium hydroxide (6.1 L, 30.5 mol, 5.98 eq) was added via addition funnel at a rate such that the maximum internal temperature was 30 °C. The mixture was then stirred for one hour 15 minutes after which the layers were allowed to settle and the layers were separated. The organic layer was washed with 1 molar aqueous sodium hydroxide (2.1 L). The aqueous layers were then combined and back extracted with isopropyl acetate (7.6 L). The organic layers were combined and washed with saturated aqueous brine (4.1 L). The organic layer was then dried over magnesium sulfate, filtered to remove solids, and then concentrated to a total volume of 4.2 L in vacuo. Hydrogen chloride in isopropyl alcohol (5.7 N, 0.90 L, 5.13 mol, 1 eq) was then added over 50 minutes using an external water/ice bath to keep the internal temperature less than 30 °C. After stirring for 45 minutes the solids were collected by filtration and the filtercake washed two times with isopropyl acetate, 2.3 L each wash. The filtercake was then partially dried and then taken forward to step 3 as a wetcake.
Step 3: Crude (5S.lRVl-(2-naphthyl -3-azabicvclof3.1.01hexane HC1 salt hot slurry in isopropyl alcohol
[00111] The wetcakes from two separate runs of step 2 (total of 4646.6 g starting amine tosylate salt) were combined and suspended in isopropyl alcohol (34.6 L) in a 50 L 3- neck round bottom flask equipped with overhead stirrer, heating mantel, thermocouple, reflux condenser, nitrogen inlet, and drying tube. The slurry was then heated to reflux, stirred for three hours at reflux, and then allowed to cool to room temperature. The solids were collected by filtration and the filtercake washed twice with isopropyl alcohol, 6.9 L each wash. The filtercake was then dried to a constant weight to give 2009.2 g of (5S,1R)-1- (2-naphthyl)-3-azabicyclo[3.1.0]hexane HCl salt (70 % yield from 4646.6 g of amine tosylate salt).
Step 4: Recrvstallization of (5S.lRVl-(2-naDhthvn-3-a2abicvclor3.1.01hexane HCl salt from ethanol to upgrade the enantiomeric excess
[00112] The (5S,lR)-l-(2-naphthyl)-3-azabicyclo[3.1.0]hexane HCl salt from step 3 (2009.2 g, 8.18 mol) was charged to a 50 L 3-neck round bottom flask equipped with an overhead stirrer, heating mantel, reflux condenser, nitrogen inlet, thermocouple, and drying tube. Ethanol (21.5 L of special industrial) was then added and the mixture heated to reflux to dissolve all solids. After dissolution of solids heating was discontinued and the mixture was allowed to cool to room temperature during which time solids reformed. The solids were then collected by filtration and the filtercake washed with ethanol (4.3 L). The filtercake was then dried to a constant weight to give 1434.6 g (71 % yield ) of recrystallized (5S,lR)-l-(2-naphthyl)-3-azabicyclo[3.1.0]hexane HCl salt. Chiral HPLC assay showed an enantiomeric excess of > 99.5 %.
Step 5: Rework to improve color profile
[00113] (5S,lR)-l-(2-naphthyl)-3-azabicyclo[3.1.0]hexane HCl (1405.6 g, 5.72 mol) was charged to a 22 L 3-neck round bottom flask equipped with overhead stirrer, heating mantel, thermocouple, nitrogen inlet and drying tube. Water (14.0 L) was added and the mixture heated to 34 °C to dissolve all solids. The solution was then transferred to a large separatory funnel and teti^ydrofuran (2.8 L) followed by isopropyl acetate (2.8 L) was added. The two phase mixture was agitated and the layers were then allowed to settle. The upper organic layer was discarded. Aqueous ammonia (1.14 L) was then added and the aqueous layer extracted with isopropylacetate (14.0 L). The organic layer was dried over magnesium sulfate, filtered, and concentrated in vacuo to give an off-white solid. The solid was dissolved in isopropyl alcohol (14.0L) and transferred to a 22 L 3-neck round bottom flask equipped with overhead stirrer, thermocouple, addition funnel, nitrogen inlet and drying tube. Hydrogen chloride in isopropyl alcohol (5.7 N, 175 mL, 1.0 mol) was then added over 10 minutes. Near the end of this addition the formation of solids was evident. The slurry was stirred for 30 minutes then additional hydrogen chloride in isopropanol (840 mL, 4.45 mol) was added over 65 minutes keeping the internal temperature less than 25 °C. The solids were collected by filtration and the filtercake washed twice with isopropyl alcohol, 2.8 L each wash. The filtercake was then dried to a constant weight to give 1277.1 g (91% yield) of the product as an off-white solid.
PATENTS
(lR,5S)-l-(naphthalen-2-yl)-3-azabicyclo[3.1.0]hexane, also known as (+)-l- (naphthalen-2-yl)-3-azabicyclo[3.1.0]hexane, is a compound useful as an unbalanced triple reuptake inhibitor (TRI), most potent towards norepinephrine reuptake (NE), one-sixth as potent towards dopamine reuptake (DA), and one-fourteenth as much towards serotonin reuptake (5- HT). This compound and its utility are disclosed in more detail in U.S. Patent Publication No. 2007/0082940, the contents of which are hereby incorporated by reference in their entirety
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US20050096395 * | Feb 12, 2003 | May 5, 2005 | Rao Srinivas G. | Methods of treating attention deficit/hyperactivity disorder (adhd) |
| US20070082940 * | Jul 25, 2006 | Apr 12, 2007 | Phil Skolnick | Novel 1-aryl-3-azabicyclo[3.1.0]hexanes: preparation and use to treat neuropsychiatric disorders |
| Reference | ||
|---|---|---|
| 1 | * | See also references of EP2819516A4 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015089111A1 * | Dec 9, 2014 | Jun 18, 2015 | Neurovance, Inc. | Novel methods |
| WO2015102826A1 * | Dec 9, 2014 | Jul 9, 2015 | Neurovance, Inc. | Novel compositions |
| US9133159 * | Apr 3, 2013 | Sep 15, 2015 | Neurovance, Inc. | 1-heteroaryl-3-azabicyclo[3.1.0]hexanes, methods for their preparation and their use as medicaments |
| US9205074 | Sep 23, 2014 | Dec 8, 2015 | Neurovance, Inc. | 1-aryl-3-azabicyclo[3.1.0]hexanes: preparation and use to treat neuropsychiatric disorders |
| US20160303076 * | Dec 9, 2014 | Oct 20, 2016 | Neurovance, Inc. | Novel methods |
References
- Jump up^ “3-Neurotransmitters, 1-Molecule: Optimized Ratios”. Neurovance.
- Jump up^ “EB-1020, a Non-Stimulant Norepinephrine and Dopamine – Preferring Reuptake Inhibitor for the Treatment of Adult ADHD” (PDF). Neurovance.
- Jump up^ Frank P. Bymaster; Krystyna Golembiowska; Magdalena Kowalska; Yong Kee Choi; Frank I. Tarazi (June 2012). “Pharmacological characterization of the norepinephrine and dopamine reuptake inhibitor EB-1020: Implications for treatment of attention-deficit hyperactivity disorder”. Synapse. 66 (6): 522–532. doi:10.1002/syn.21538. PMID 22298359.
External links
 |
|
| Legal status | |
|---|---|
| Legal status |
|
| Identifiers | |
| CAS Number | 924012-43-1 |
| PubChem (CID) | 16095349 |
| ChemSpider | 17253639 |
| Chemical and physical data | |
| Formula | C15H15N |
| Molar mass | 209.28 g/mol |
| 3D model (Jmol) | Interactive image |
///////CENTANAFADINE, PHASE 2, UNII-D2A6T4UH9C, EB-1020, D2A6T4UH9C, 924012-43-1, CTN SR, EB-1020, EB-1020 SR,
C1C2C1(CNC2)C3=CC4=CC=CC=C4C=C3
