















Copanlisib, BAY 80-6946,
- BAY 84-1236
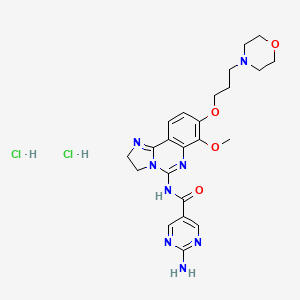
- Molecular FormulaC23H28N8O4
- Average mass480.520 Da
Cas 1032568-63-0 [RN]
1402152-26-4 MONO HCL
UNII-WI6V529FZ9
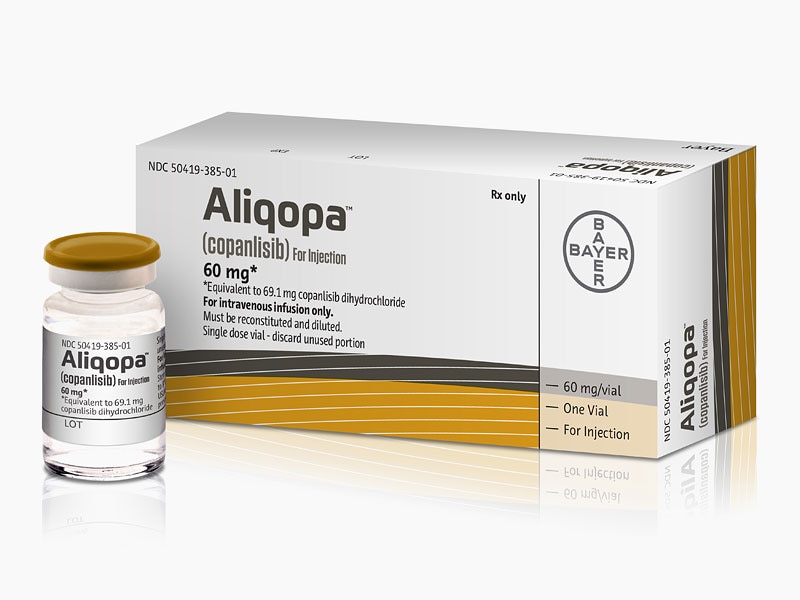
FDA Approved September 2017
Copanlisib (BAY 80-6946), developed by Bayer, is a selective Class I phosphoinositide 3-kinase inhibitor[1] which has shown promise in Phase I/II clinical trials for the treatment of non-Hodgkin lymphoma and chronic lymphocytic leukemia.[2]
![]()
Copanlisib is a selective pan-Class I phosphoinositide 3-kinase (PI3K/Phosphatidylinositol-4,5-bisphosphate 3-kinase/phosphatidylinositide 3-kinase) inhibitor that was first developed by Bayer Healthcare Pharmaceuticals, Inc. The drug targets the enzyme that plays a role in regulating cell growth and survival. Copanlisib was granted accelerated approval on September 14, 2017 under the market name Aliqopa for the treatment of adult patients with relapsed follicular lymphoma and a treatment history of at least two prior systemic therapies. Follicular lymphoma is a slow-growing type of non-Hodgkin lymphoma that is caused by unregulated proliferation and growth of lymphocytes. The active ingredient in Aliquopa intravenous therapy is copanlisib dihydrochloride.

1402152-46-8 CAS X=4,
1919050-77-3 CAS X=1
The FDA awarded copanlisib orphan drug status for follicular lymphoma in February 2015.[3]
Phase II clinical trials are in progress for treatment of endometrial cancer,[4] diffuse large B-cell lymphoma,[5] cholangiocarcinoma,[6]and non-Hodgkin lymphoma.[7] Copanlisib in combination with R-CHOP or R-B (rituximab and bendamustine) is in a phase III trial for relapsed indolent non-Hodgkin lymphoma (NHL).[8] Two separate phase III trials are investigating the use of copanlisib in combination with rituximab for indolent NHL[9] and the other using copanlisib alone in cases of rituximab-refractory indolent NHL.[10]
Copanlisib hydrochloride, a phosphatidylinositol 3-Kinase inhibitor developed by Bayer, was first approved and launched in 2017 in the U.S. for the intravenous treatment of adults with relapsed follicular lymphoma who have received at least two prior treatments.
In 2015, orphan drug designation was assigned in the U.S. for the treatment of follicular lymphoma. In 2017, additional orphan drug designations were granted in the U.S. for the treatment of splenic, nodal and extranodal marginal zone lymphoma.
SYN
WO 2017049983
PATENTS
| Inventors | Martin Hentemann, Jill Wood, William Scott, Martin Michels, Ann-Marie Campbell, Ann-Marie Bullion, R. Bruce Rowley, Aniko Redman, Less « |
| Applicant | Bayer Schering Pharma Aktiengesellschaft |
Example 13
Preparation of 2-amino-N-r7-methoxy-8-(3-morpholin-4-ylpropoxy)-2.3- dihvdroimidazori^-clquinazolin-S-vHpvrimidine-S-carboxamide.

Step 1 : Preparation of 4-hvdroxy-3-methoxy-2-nitrobenzonitrile

4-Hydroxy-3-methoxy-2-nitrobenzaldehyde (200 g, 1.01 mol) was dissolved in THF (2.5 L) and then ammonium hydroxide (2.5 L) was added followed by iodine (464 g, 1.8 mol). The resulting mixture was allowed to stir for 2 days at which time it was concentrated under reduced pressure. The residue was acidified with HCI (2 N) and extracted into diethyl ether. The organic layer was washed with brine and dried (sodium sulfate) and concentrated under reduced pressure. The residue was washed with diethyl ether and dried under vacuum to provide the title compound (166 g, 84%): 1H NMR (DMSO-cfe) δ: 11.91 (1 H, s), 7.67 (1 H, d), 7.20 (1 H, d), 3.88 (3H, s)
Step 2: Preparation of 3-methoxy-4-(3-morpholin-4-ylpropoxy)-2-nitrobenzonitrile

To a solution of 4-hydroxy-3-methoxy-2-nitrobenzonitrile (3.9 g, 20.1 mmol) in DMF (150 mL) was added cesium carbonate (19.6 g, 60.3 mmol) and Intermediate C (5.0 g, 24.8 mmol). The reaction mixture was heated at 75 0C overnight then cooled to room temperature and filtered through a pad of silica gel and concentrated under reduced pressure. The material thus obtained was used without further purification
Step 3: Preparation of 2-amino-3-methoxy-4-(3-morpholin-4-ylpropoxy)benzonitrile
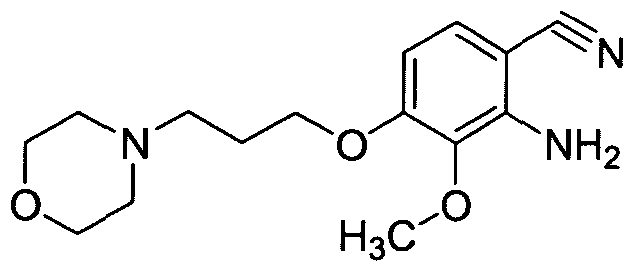
3-Methoxy-4-(3-morpholin-4-ylpropoxy)-2-nitrobenzonitrile (7.7 g, 24.1 mmol) was suspended in acetic acid (170 ml_) and cooled to 0 °C. Water (0.4 ml_) was added, followed by iron powder (6.7 g, 120 mmol) and the resulting mixture was stirred at room temperature for 4 h at which time the reaction mixture was filtered through a pad of Celite and washed with acetic acid (400 ml_). The filtrate was concentrated under reduced pressure to 100 mL and diluted with EtOAc (200 ml.) at which time potassium carbonate was added slowly. The resulting slurry was filtered through a pad of Celite washing with EtOAc and water. The layers were separated and the organic layer was washed with saturated sodium bicarbonate solution. The organic layer was separated and passed through a pad of silica gel. The resultant solution was concentrated under reduced pressure to provide the title compound (6.5 g, 92%): 1H NMR (DMSO-Cf6) δ: 7.13 (1 H1 d), 6.38 (1 H, d), 5.63 (2H1 br s), 4.04 (2H, t), 3.65 (3H, s), 3.55 (4H1 br t), 2.41 (2H, t), 2.38 (4H1 m), 1.88 (2H1 quint.).
Step 4: Preparation of 6-(4.5-dihvdro-1 H-imidazol-2-v0-2-methoxy-3-(3-morpholin- 4-ylpropoxy)aniline

To a degassed mixture of 2-amino-3-methoxy-4-(3-morpholin-4-ylpropoxy)benzonitrile (6.5 g, 22.2 mmol) and ethylene diamine (40 mL) was added sulfur (1.8 g, 55.4 mmol). The mixture was stirred at 100 °C for 3 h at which time water was added to the reaction mixture. The precipitate that was formed was collected and washed with water and then dried overnight under vacuum to provide the title compound (3.2 g, 43%): HPLC MS RT = 1.25 min, MH+= 335.2; 1H NMR (DMSO-Cf6) δ: 7.15 (1H, d), 6.86 (2H, br s), 6.25 (1 H, d), 4.02 (2H, t), 3.66 (3H, s), 3.57 (8H, m), 2.46 (2H, t), 2.44 (4H, m), 1.89 (2H, quint.). Step 5: Preparation of 7-methoxy-8-(3-morpholin-4-ylpropoxy)-2.3- dihvdroimidazof1.2-clquinazolin-5-amine

Cyanogen bromide (10.9 g, 102.9 mmol) was added to a mixture of 6-(4,5-dihydro-1 H- imidazol-2-yl)-2-methoxy-3-(3-morpholin-4-ylpropoxy)aniline (17.2 g, 51.4 mmol) and TEA (15.6 g, 154.3 mmol) in DCM (200 ml_) precooled to 0 0C. After 1 h the reaction mixture was concentrated under reduced pressure and the resulting residue stirred with EtOAc (300 mL) overnight at rt. The resulting slurry was filtered to generate the title compound contaminated with triethylamine hydrobromide (26.2 g, 71%): HPLC MS RT = 0.17 min, MH+= 360.2.
Step 6: Preparation of 2-amino-N-r7-methoxy-8-(3-morpholin-4-ylpropoxy)-2.3- dihvdroimidazori ^-clquinazolin-S-vnpyrimidine-δ-carboxamide.

7-Methoxy-8-(3-morpholin-4-ylpropoxy)-2,3-dihydroimidazo[1 ,2-c]quinazolin-5-amine (100 mg, 0.22 mol) was dissolved in DMF (5 mL), and Intermediate B (46 mg, 0.33 mmol) was added. PYBOP (173 mg, 0.33 mmol) and diisopropylethylamine (0.16 mL, 0.89 mmol) were subsequently added, and the mixture was stirred at rt overnight. EtOAc was added, and the solids were isolated by vacuum filtration to give the title compound (42.7 mg, 40%): HPLC MS RT = 1.09 min, MH+= 481.2; 1H NMR (DMSO-Cf6 + 2 drops TFA-tf) δ: 9.01 (2H, s), 8.04 (1 H, d), 7.43 (1 H, d), 4.54 (2H, m), 4.34 (2H, br t), 4.23 (2H, m), 4.04 (2H, m), 4.00 (3H, s), 3.65 (2H, br t), 3.52 (2H, m), 3.31 (2H, m), 3.18 (2H, m), 2.25 (2H, m).
PATENT
TRANSLATED
Example VI:
[0053] a nitrogen atmosphere, the reaction flask was added 7-methoxy-8- (3-morpholin-4-yl-propoxy) -2,3-dihydro-imidazo [l, 2-c] quinoline tetrazol-5-amine (V) (0 • 36g, lmmol), 2- amino-5-carboxylic acid (0 • 15g, l.lmmol) and acetonitrile 25mL, condensing agent added benzotriazole-1-yl yloxy-tris (dimethylamino) phosphonium hexafluorophosphate key (0.49g, 1. lmmol) and the base catalyst 1,5_-diazabicyclo [4. 3.0] – non-5-ene (0 . 50g, 4mmol), at room temperature for 12 hours.Then heated to 50-60 ° C, the reaction was stirred for 6-8 hours, TLC the reaction was complete. The solvent was distilled off under reduced pressure, cooled to room temperature, ethyl acetate was added solid separated. Filter cake washed with cold methanol and vacuum dried to give an off-white solid Kupannixi (1) 0.278, showing a yield of 56.3% -] \ ^ 111/2: 481 [] \ 1+ buckle + 1 111 bandit ? (square) (: 13). 5 2.05 (111,211), 2.48 (111,411), 2. 56 (m, 2H), 3 72 (t, 4H), 4 02 (s, 3H),. 4. 16 (m, 7H), 5. 36 (s, 2H), 6. 84 (d, 1H), 7. 08 (d, 1H), 9. 10 (s, 2H) square
PATENT
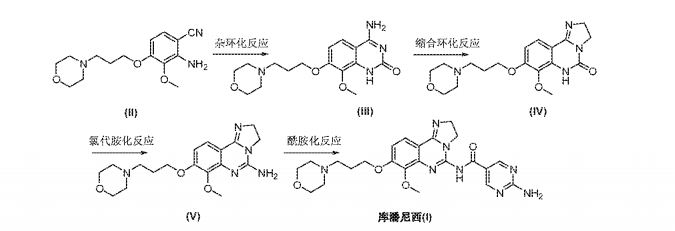
2-amino-N-[7-methoxy-8-(3-morpholin-4-ylpropoxy)-2,3-dihydroimidazo[1 ,2-c]quinazolin-5-yl]pyrimidine-5-carboxamide (10), (which is hereinafter referred to as„copanlisib”), is a proprietary cancer agent with a novel mechanism of action, inhibiting Class I phosphatidylinositol-3-kinases (PI3Ks). This class of kinases is an attractive target since PI3Ks play a central role in the transduction of cellular signals from surface receptors for survival and proliferation. Copanlisib exhibits a broad spectrum of activity against tumours of multiple histologic types, both in vitro and in vivo.
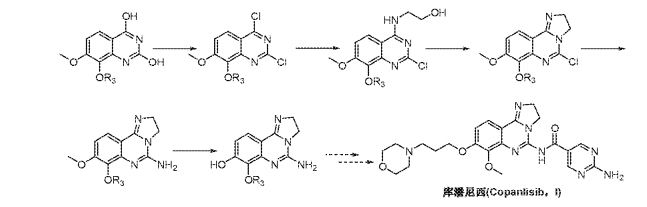
Copanlisib may be synthesised according to the methods given in international patent application PCT/EP2003/010377, published as WO 04/029055 A1 on April 08, 2004, (which is incorporated herein by reference in its entirety), on pp. 26 et seq.
Copanlisib is published in international patent application PCT/US2007/024985, published as WO 2008/070150 A1 on June 12, 2008, (which is incorporated herein by reference in its entirety), as the compound of Example 13 : 2-amino-N-[7-methoxy-8-(3-morpholin-4-ylpropoxy)-2,3-dihydroimidazo[1 ,2-c]quinazolin-5-yl]pyrimidine-5-carboxamide.
Copanlisib may be synthesized according to the methods given in WO 2008/070150, pp. 9 et seq., and on pp. 42 et seq. Biological test data for said compound of formula (I) is given in WO 2008/070150 on pp. 101 to 107.
2-amino-N-[7-methoxy-8-(3-morpholin-4-ylpropoxy)-2,3-dihydroimid-azo[1 ,2-c]quinazolin-5-yl]pyrimidine-5-carboxamide dihydrochloride (1 1 ), (which is hereinafter referred to as „copanlisib dihydrochloride”) is published in international patent application PCT/EP2012/055600, published as WO 2012/136553 on October 1 1 , 2012, (which is incorporated herein by reference in its entirety), as the compound of Examples 1 and 2 : 2-amino-N-[7-methoxy-8-(3-morpholin-4-ylpropoxy)-2,3-dihydroimidazo[1 ,2-c]quinazolin-5-yl]pyrimidine-5-carboxamide dinydrochloride : it may be synthesized according to the methods given in said Examples 1 and 2.
Copanlisib may exist in one or more tautomeric forms : tautomers, sometimes referred to as proton-shift tautomers, are two or more compounds that are related by the migration of a hydrogen atom accompanied by the migration of one or more single bonds and one or more adjacent double bonds.
Copanlisib may for example exist in tautomeric form (la), tautomeric form (lb), or tautomeric form (Ic), or may exist as a mixture of any of these forms, as depicted below. It is intended that all such tautomeric forms are included within the scope of the present invention.



Copanlisib may exist as a solvate : a solvate for the purpose of this invention is a complex of a solvent and copanlisib in the solid state. Exemplary solvates include, but are not limited to, complexes of copanlisib with ethanol or methanol.
Copanlisib and copanlisib dihydrochloride may exist as a hydrate. Hydrates are a specific form of solvate wherein the solvent is water, wherein said water is a structural element of the crystal lattice of copanlisib or of copanlisib dihydrochloride. It is possible for the amount of said water to exist in a stoichiometric or non-stoichiometric ratio. In the case of stoichiometric hydrates, a hemi-, (semi-), mono-, sesqui-, di-, tri-, tetra-, or penta-hydrate of copanlisib or of copanlisib dihydrochloride is possible. It is also possible for water to be present on the surface of the crystal lattice of copanlisib or of copanlisib dihydrochloride. The present invention includes all such hydrates of copanlisib or of copanlisib dihydrochloride, in particular copanlisib dihydrochloride hydrate referred to as “hydrate I”, as prepared and characterised in the experimental section herein, or as “hydrate II”, as prepared and characterised in the experimental section herein.
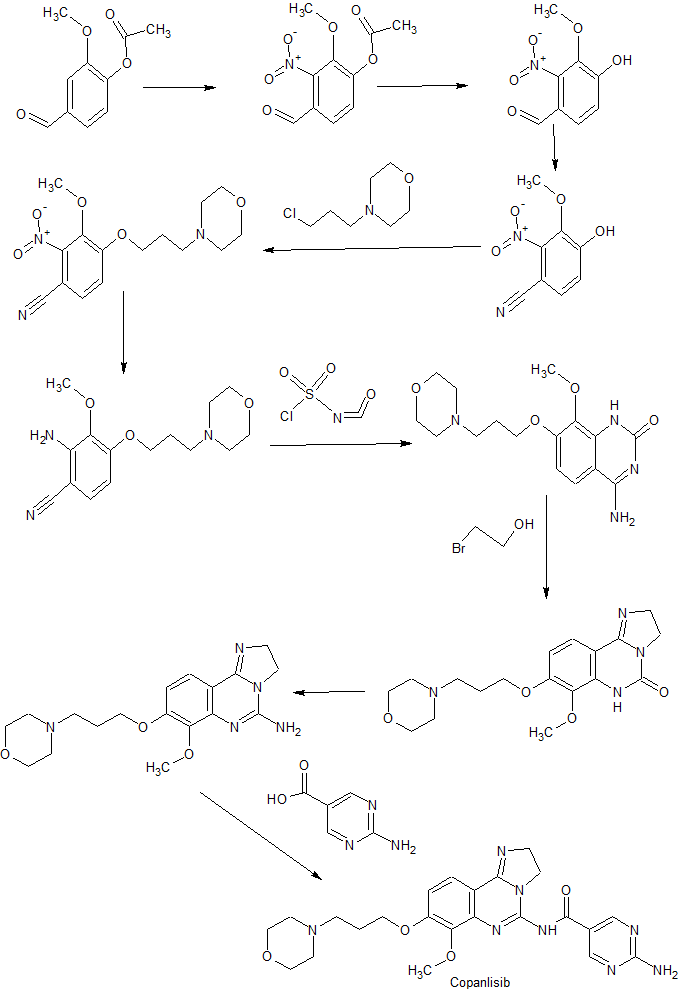
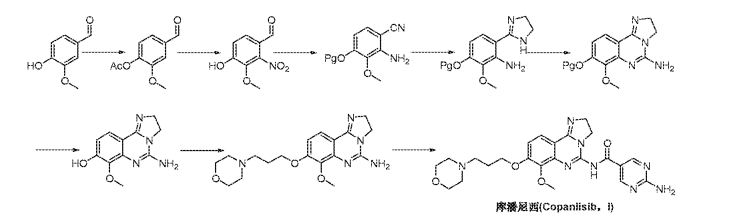
As mentioned supra, copanlisib is, in WO 2008/070150, described on pp. 9 et seq., and may be synthesized according to the methods given therein on pp. 42 et seq., viz. :
Reaction Scheme 1 :


(I)
In Reaction Scheme 1 , vanillin acetate can be converted to intermediate (III) via nitration conditions such as neat fuming nitric acid or nitric acid in the presence of another strong acid such as sulfuric acid. Hydrolysis of the acetate in intermediate (III) would be expected in the presence of bases such as sodium
hydroxide, lithium hydroxide, or potassium hydroxide in a protic solvent such as methanol. Protection of intermediate (IV) to generate compounds of Formula (V) could be accomplished by standard methods (Greene, T.W.; Wuts, P.G.M.; Protective Groups in Organic Synthesis; Wiley & Sons: New York, 1999). Conversion of compounds of formula (V) to those of formula (VI) can be achieved using ammonia in the presence of iodine in an aprotic solvent such as THF or dioxane. Reduction of the nitro group in formula (VI) could be accomplished using iron in acetic acid or hydrogen gas in the presence of a suitable palladium, platinum or nickel catalyst. Conversion of compounds of formula (VII) to the imidazoline of formula (VIII) is best accomplished using ethylenediamine in the presence of a catalyst such as elemental sulfur with heating. The cyclization of compounds of formula (VIII) to those of formula (IX) is accomplished using cyanogen bromide in the presence of an amine base such as triethylamine, diisopropylethylamine, or pyridine in a halogenated solvent such as DCM or dichloroethane. Removal of the protecting group in formula (IX) will be dependent on the group selected and can be accomplished by standard methods (Greene, T.W.; Wuts, P.G.M.; Protective Groups in Organic Synthesis; Wiley & Sons: New York, 1999). Alkylation of the phenol in formula (X) can be achieved using a base such as cesium carbonate, sodium hydride, or potassium t-butoxide in a polar aprotic solvent such as DMF or DMSO with introduction of a side chain bearing an appropriate leaving group such as a halide, or a sulfonate group. Lastly, amides of formula (I) can be formed using activated esters such as acid chlorides and anhydrides or alternatively formed using carboxylic acids and appropriate coupling agents such as PYBOP, DCC, or EDCI in polar aprotic solvents.
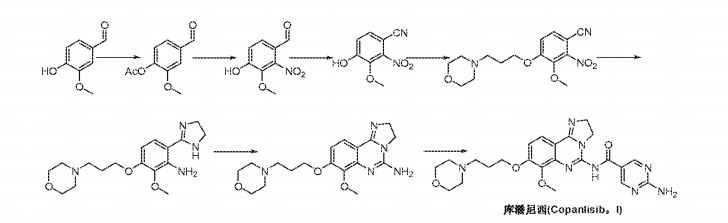
Reaction Scheme 2 :

Reaction Scheme 3



Step A9: N-[3-(dimethylamino)propyl]-N’-ethylcarbodiimide hydrochloride (“EDCI”) is used as coupling reagent. Copanlisib is isolated by simple filtration.
Step A1 1 : Easy purification of copanlisib via its dihydrochloride
(dihydrochloride is the final product)
Hence, in a first aspect, the present invention relates to a method of preparing copanlisib (10) via the following steps shown in Reaction Scheme 3, infra :
Reaction Scheme 3 : 


Example 1 : Step A1 : Preparation of 4-acetoxy-3-methoxy-2-nitrobenzaldehyde (2)
3.94 kg of nitric acid (65 w%) were added to 5.87 kg of concentrated sulfuric acid at 0°C (nitrating acid). 1 .5 kg of vanillin acetate were dissolved in 2.9 kg of dichloromethane (vanillin acetate solution). Both solutions reacted in a micro reactor with flow rates of app. 8.0 mL/min (nitrating acid) and app. 4.0 mL/min (vanillin acetate solution) at 5°C. The reaction mixture was directly dosed into 8 kg of water at 3°C. After 3h flow rates were increased to 10 mL/min (nitrating acid) and 5.0 mL/min (vanillin acetate solution). After additional 9 h the flow reaction was completed. The layers were separated at r.t., and the aqueous phase was extracted with 2 L of dichloromethane. The combined organic phases were washed with 2 L of saturated sodium bicarbonate, and then 0.8 L of water. The dichloromethane solution was concentrated in vacuum to app. 3 L, 3.9 L of methanol were added and app. the same volume was removed by distillation again. Additional 3.9 L of methanol were added, and the solution concentrated to a volume of app. 3.5 L. This solution of 4-acetoxy-3-methoxy-2-nitrobenzaldehyde (2) was directly used in the next step.
Example 2 : Step A2 : Preparation of 4-hydroxy -3-methoxy-2-nitrobenzaldehyde (2-nitro-vanillin) (3)
To the solution of 4-acetoxy-3-methoxy-2-nitrobenzaldehyde (2) prepared as described in example 1 (see above) 1 .25 kg of methanol were added, followed by 2.26 kg of potassium carbonate. The mixture was stirred at 30°C for 3h. 7.3 kg of dichloromethane and 12.8 kg of aqueous hydrochloric acid (10 w%) were added at < 30°C (pH 0.5 – 1 ). The mixture was stirred for 15 min, and the layers were separated. The organic layer was filtered, and the filter cake washed with 0.5 L of dichloromethane. The aqueous layer was extracted twice with 4.1 kg of
dichloromethane. The combined organic layers were concentrated in vacuum to app. 4 L. 3.41 kg of toluene were added, and the mixture concentrated to a final volume of app. 4 L. The mixture was cooled to 0°C. After 90 min the suspension was filtered. The collected solids were washed with cold toluene and dried to give 0.95 kg (62 %).
1H-NMR (400 MHz, de-DMSO): δ =3.84 (s, 3H), 7.23 (d, 1 H), 7.73 (d, 1 H), 9.74 (s, 1 H), 1 1 .82 (brs, 1 H).
NMR spectrum also contains signals of regioisomer 6-nitrovanillin (app. 10%): δ = 3.95 (s, 3H), 7.37 (s, 1 H), 7.51 (s, 1 H), 10.16 (s, 1 H), 1 1 .1 1 (brs, 1 H).
Example 3 : Step A3 : Preparation of 4-(benzyloxy)-3-methoxy-2-nitrobenzaldehyde (4) :
10 g of 3 were dissolved in 45 mL DMF at 25 °C. This solution was charged with 14 g potassium carbonate and the temperature did rise to app. 30 °C. Into this suspension 7.1 mL benzyl bromide was dosed in 15minutes at a temperature of 30 °C. The reaction mixture was stirred for 2 hours to complete the reaction. After cooling to 25 °C 125 mL water was added. The suspension was filtered, washed twice with 50 mL water and once with water / methanol (10 mL / 10 mL) and tried at 40 °C under reduced pressure. In this way 14.2 g (97% yield) of 4 were obtained as a yellowish solid.
1 H-NMR (500 MHz, d6-DMSO): 3.86 (s, 3H); 5.38 (s, 2 H); 7.45 (m, 5H); 7.62 (d, 2H); 7.91 (d, 2H); 9.81 (s, 1 H).
Example 4a : Step A4 : 2-[4-(benzyloxy)-3-methoxy-2-nitrophenyl]-4,5-dihydro-1 H-imidazole (5) : Method A
10 g of 4 were dissolved in 100 mL methanol and 2.5 g ethylenediamine were added at 20-25 °C. The reaction mixture was stirred at this temperature for one hour, cooled to 0°C and a solution of N- bromosuccinimide (8.1 g) in 60 mL
acetonitrile was added. Stirring was continued for 1 .5 h and the reaction mixture was warmed to 20 °C and stirred for another 60 minutes. The reaction was quenched with a solution of 8.6 g NaHCO3 and 2.2 g Na2SO3 in 100 mL water. After 10 minutes 230 mL water was added, the product was filtered, washed with 40 mL water and tried at 40 °C under reduced pressure. In this way 8.9 g (78% yield) of 5 was obtained as an white solid.
1 H-NMR (500 MHz, d6-DMSO): 3.31 (s, 4H); 3.83 (s, 3H); 5.29 (s, 2 H); 6.88 (s, 1 H); 7.37 (t, 1 H); 7.43 (m, 3H); 7.50 (m, 3H).
Example 4b : Step A4 : 2-[4-(benzyloxy)-3-methoxy-2-nitrophenyl]-4,5-dihydro-1 H-imidazole (5) : Method B
28.7 kg of compound 4 were dissolved in 231 kg dichloromethane at 20 °C and 8.2 kg ethylenediamine were added. After stirring for 60 minutes N-bromosuccinimide was added in 4 portions (4 x 5.8 kg) controlling that the temperature did not exceed 25°C. When the addition was completed stirring was continued for 90 minutes at 22 °C. To the reaction mixture 9 kg potassium carbonate in 39 kg water was added and the layers were separated. From the organic layer 150 kg of solvent was removed via distillation and 67 kg toluene was added. Another 50 kg solvent was removed under reduced pressure and 40 kg toluene was added. After stirring for 30 minutes at 35-45 °C the reaction was cooled to 20 °C and the product was isolated via filtration. The product was washed with toluene (19 kg), tried under reduced pressure and 26.6 kg (81 % yield) of a brown product was obtained.
Example 5 : Step A5 : 3-(benzyloxy)-6-(4,5-dihydro-1 H-imidazol-2-yl)-2-methoxyaniline (6) :
8.6 g of compound 5 were suspended in 55 mL THF and 1 .4 g of 1 %Pt/0.2% Fe/C in 4 mL water was added. The mixture was heated to 45 °C and hydrogenated at 3 bar hydrogen pressure for 30 minutes. The catalyst was
filtered off and washed two times with THF. THF was removed via distillation and 65 mL isopropanol/water 1/1 were added to the reaction mixture. The solvent remaining THF was removed via distillation and 86 mL isopropanol/water 1/1 was added. The suspension was stirred for one hour, filtered, washed twice with isopropanol/water 1/1 and dried under reduced pressure to yield 7.8g (99% yield) of an white solid.
1 H-NMR (500 MHz, d6-DMSO): 3.26 (t, 2H); 3.68 (s, 3H); 3.82 (t, 2H); 5.13 (s, 2 H); 6.35 (d, 1 H); 6.70 (s, 1 H); 6.93 (bs, 2 H); 7.17 (d, 1 H); 7.33 (t, 1 H); 7.40 (t, 2H); 7.45 (d, 2H).
Example 6a : Step A6 : 8-(benzyloxy)-7-methoxy-2,3-dihydroimidazo[1 ,2-c]quinazolin-5-amine (7) : Method A
10 g of 6 were suspended in 65 mL acetonitrile and 6.1 mL triethylamine were added. At 5-10 °C 8.4 mL bromocyanide 50% in acetonitrile were added over one hour and stirring was continued for one hour. 86 mL 2% NaOH were added and the reaction mixture was heated to 45 °C and stirred for one hour. The suspension was cool to 10 °C, filtered and washed with water/acetone 80/20. To further improve the quality of the material the wet product was stirred in 50 mL toluene at 20-25 °C. The product was filtered off, washed with toluene and dried under reduced pressure. In this way 8.8 g (81 % yield) of 7 was isolated as a white solid.
1 H-NMR (500 MHz, d6-DMSO): 3.73 (s, 3H); 3.87 (m, 4H); 5.14 (s, 2 H); 6.65 (bs, 2 H); 6.78 (d, 1 H); 7.33 (m, 1 H); 7.40 (m, 3 H); 7.46 (m, 2H).
Example 6b : Step A6 : 8-(benzyloxy)-7-methoxy-2,3-dihydroimidazo[1 ,2-c]quinazolin-5-amine (8) : Method B
20 kg of compound 6 were dissolved in 218 kg dichloromethane at 20 °C and the mixture was cooled to 5 °C. At this temperature 23.2 kg triethylamine was dosed in 15 minutes and subsequently 25.2 kg bromocyanide (3 M in
dichloromethane) was dosed in 60 minutes to the reaction mixture. After stirring for one hour at 22 °C the reaction was concentrated and 188 kg of solvent were removed under reduced pressure. Acetone (40 kg) and water (50 kg) were added and another 100 kg of solvent were removed via distillation. Acetone (40 kg) and water (150 kg) were added and stirring was continued for 30 minutes at 36°C. After cooling to 2 °C the suspension was stirred for 30 minutes, isolated, washed with 80 kg of cold water and tried under reduced pressure. With this procedure 20.7 kg (95% yield) of an off-white product was obtained.
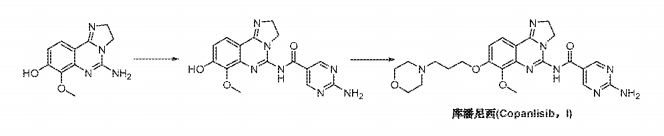
Example 7a : Step A7 : Method A: preparation of 5-amino-7-methoxy-2,3-dihydroimidazo[1 ,2-c]quinazolin-8-ol (8) :
A mixture of 2 kg of 8-(benzyloxy)-7-methoxy-2,3-dihydroimidazo[1 ,2-c]quinazolin-5-amine, 203 g of 5% Palladium on charcoal (50% water wetted) and 31 .8 kg of Ν,Ν-dimethylformamide was stirred at 60°C under 3 bar of hydrogen for 18 h. The mixture was filtered, and the residue was washed with 7.5 kg of Ν,Ν-dimethylformamide. The filtrate (38.2 kg) was concentrated in vacuum (ap. 27 L of distillate collected and discarded). The remaining mixture was cooled from 50°C to 22°C within 1 h, during this cooling phase 14.4 kg of water were added within 30 min. The resulting suspension was stirred at 22°C for 1 h and then filtered. The collected solids were washed with water and dried in vacuum to yield 0.94 kg (65 %).
1H-NMR (400 MHz, de-DMSO): δ = 3.72 (s, 3H), 3.85 (m, 4H), 6.47 (d, 1 H), 6.59 (bs, 1 H), 7.29 (d, 1 H), 9.30 (bs, 1 H).
Example 7b : Step A7 Method B : preparation of 5-amino-7-methoxy-2,3-dihydroimidazo[1 ,2-c]quinazolin-8-ol (8) :
222.8 g of trifluroacetic acid were added to a mixture of 600 g of 8-(benzyloxy)-7-methoxy-2,3-dihydroimidazo[1 ,2-c]quinazolin-5-amine and 2850 g of DMF. 18 g of 5% Palladium on charcoal (50% water wetted) were added. The mixture
was stirred at under 3 bar of hydrogen overnight. The catalyst was removed by filtration and washed with 570 g of DMF. The filtrate was concentrated in vacuum (432 g of distillate collected and discarded). 4095 ml of 0.5 M aqueous sodium hydroxide solution was added within 2 hours. The resulting suspension was stirred overnight. The product was isolated using a centrifuge. The collected solids were washed with water. The isolated material (480.2g; containing app. 25 w% water) can be directly used in the next step (example 8b).
Example 8a : Step A8 : Method A : preparation of 7-methoxy-8-[3-(morpholin-4-yl)propoxy]-2,3-dihydroimidazo[1 ,2-c]quinazolin-5-amine (9) :
2.5 kg of potassium carbonate were added to a mixture of 1 .4 kg of 5-amino-7-methoxy-2,3-dihydroimidazo[1 ,2-c]quinazolin-8-ol, 14 L of n-butanol, 1 .4 L of Ν,Ν-dimethylformamide and 1 .4 L of water. 1 .57 kg of 4-(3-chloropropyl)morpholine hydrochloride were added. The resulting suspension was heated to 90°C and stirred at this temperature for 5 h. The mixture was cooled to r.t.. At 50°C 8.4 kg of water were added. The mixture was stirred at r.t. for 15 min. After phase separation the aqueous phase was extracted with 12 L of n-butanol. The combined organic phases were concentrated in vacuum to a volume of ap. 1 1 L. 10.7 L of terf-butyl methyl ether were added at 50°C. The resulting mixture was cooled within 2 h to 0°C and stirred at this temperature for 1 h. The suspension was filtered, and the collected solids were washed with tert-butyl methyl ether and dried to give 1 .85 kg (86 %).
The isolated 1 .85 kg were combined with additional 0.85 kg of material produced according to the same process. 10.8 L of water were added and the mixture heated up to 60°C. The mixture was stirred at this temperature for 10 min, then cooled to 45°C within 30 min and then to 0°C within 1 h. The suspension was stirred at 0°C for 2 h and then filtered. The solids were washed with cold water and dried to yield 2.5 kg.
1H-NMR (400 MHz, de-DMSO): δ = 1 .88 (m, 4H), 2.36 (m, 4H), 2.44 (t, 2H), 3.57 (m, 4H), 3.70 (s, 3H), 3.88 (m, 4H), 4.04 (t, 2H), 6.63 (s, 2H), 6.69 (d, 1 H), 7.41 (d, 1 H).
HPLC: stationary phase: Kinetex C18 (150 mm, 3.0 mm ID, 2.6 μιτι particle size): mobile phase A: 0.5 ml_ trifluoro acetic acid / 1 L water; mobile phase B: 0.5 ml_ trifluoro acetic acid / L acetonitrile; UV detection at 256 nm; oven temperature: 40°C; injection volume: 2.0 μΙ_; flow 1 .0 mL/min; linear gradient in 4 steps: 0% B -> 6% B (20 min), 6 % B -> 16% B (5 min), 16% B -> 28 % B (5 min), 28 % B -> 80 % B (4 min), 4 minutes holding time at 80% B; purity: >99,5 % (Rt=1 1 .0 min), relevant potential by-products: degradation product 1 at RRT (relative retention time) of 0.60 (6.6 min) typically <0.05 %, 5-amino-7-methoxy-2,3-dihydroimidazo[1 ,2-c]quinazolin-8-ol RRT 0.71 (7.8 min): typically <0.05 %, degradation product 2 RRT 1 .31 (14.4 min): typically <0.05 %, 7-methoxy-5-{[3-(morpholin-4-yl)propyl]amino}-2,3-dihydroimidazo[1 ,2-c]quinazolin-8-ol RRT 1 .39 (15.3 min): typically <0.05 %, 9-methoxy-8-[3-(morpholin-4-yl)propoxy]-2,3-dihydroimidazo[1 ,2-c]quinazolin-5-amine RRT 1 .43 (15.7 min): typically <0.05 %, degradation product 3 RRT 1 .49 (16.4 min): typically <0.05 %, 7-methoxy-8-[3-(morpholin-4-yl)propoxy]-N-[3-(morpholin-4-yl)propyl]-2,3-dihydroimidazo[1 ,2-c]quinazolin-5-amine RRT 1 .51 (16.7 min): typically <0.10 %, 8-(benzyloxy)-7-methoxy-2,3-dihydroimidazo[1 ,2-c]quinazolin-5-amine RRT 2.56 (28.2 min): typically <0.05 %, 8-(benzyloxy)-7-methoxy-N-[3-(morpholin-4-yl)propyl]-2,3-dihydroimidazo[1 ,2-c]quinazolin-5-amine RRT 2.59 (28.5 min): typically <0.05 %.
Example 8b: : Step A8 (Method B): preparation of 7-methoxy-8-[3-(morpholin-4-yl)propoxy]-2,3-dihydroimidazo[1 ,2-c]quinazolin-5-amine (9) :
13.53 g of 5-amino-7-methoxy-2,3-dihydroimidazo[1 ,2-c]quinazolin-8-ol (containing app. 26 w% of water) were suspended in 1 10 g of n-butanol. The mixture was concentrated in vacuum (13.5 g of distillate collected and discarded). 17.9 g of potassium carbonate and 1 1 .2 g of 4-(3-chloropropyl)morpholine hydrochloride were added. The resulting mixture was heated to 90°C and stirred at this temperature for 4 hours. The reaction mixture was cooled to to 50°C, and 70 g of water were added. The layers were separated. The organic layer was concentrated in vacuum (54 g of distillate collected and discard). 90 g of terf-butyl methyl ether were added at 65°C. The resulting mixture was cooled to 0°C. The mixture was filtered, and the collected solids washed with terf-butyl methyl ether and then dried in vacuum to yield 13.4 g (86%).
13.1 g of the isolated material were suspended in 65.7 g of water. The mixture was heated to 60°C. The resulting solution was slowly cooled to 0°C. The precipitated solids were isolated by filtration, washed with water and dried in vacuum to yield 12.0 g (92%).
Example 9: Step A10 : Preparation of 2-aminopyrimidine-5-carboxylic acid (9b)
1 kg of methyl 3,3-dimethoxypropanoate was dissolved in 7 L of 1 ,4-dioxane. 1 .58 kg of sodium methoxide solution (30 w% in methanol) were added. The mixture was heated to reflux, and ap. 4.9 kg of distillate were removed. The resulting suspension was cooled to r.t., and 0.5 kg of methyl formate was added. The reaction mixture was stirred overnight, then 0.71 kg of guanidine hydrochloride was added, and the reaction mixture was stirred at r.t. for 2 h. The reaction mixture was then heated to reflux, and stirred for 2 h. 13.5 L of water were added, followed by 0.72 kg of aqueous sodium hydroxide solution (45 w%). The reaction mixture was heated at reflux for additional 0.5 h, and then cooled to 50°C. 0.92 kg of aqueous hydrochloric acid (25 w%) were added until pH 6 was reached. Seeding crystals were added, and additional 0.84 kg of aqueous hydrochloric acid (25 w%) were added at 50°C until pH 2 was reached. The mixture was cooled to 20°C and stirred overnight. The suspension was filtered, the collected solids washed twice with water, then twice with methanol, yielding 0.61 kg (65%).
Four batches produced according to the above procedure were combined (total 2.42 kg). 12 L of ethanol were added, and the resulting suspension was stirred at r.t. for 2.5 h. The mixture was filtered. The collected solids were washed with ethanol and dried in vacuum to yield 2.38 kg.
To 800 g of this material 2.5 L of dichloromethane and 4 L of water were added, followed by 1375 ml_ of dicyclohexylamine. The mixture was stirred for 30 min. at r.t. and filtered. The collected solids are discarded. The phases of the filtrate are separated, and the organic phase was discarded. 345 ml_ of aqueous sodium hydroxide solution (45 w%) were added to the aqueous phase. The aqueous phase was extracted with 2.5 L of ethyl acetate. The phases were separated and the organic phase discarded. The pH value of the aqueous phase was adjusted to pH 2 using app. 500 ml_ of hydrochloric acid (37 w%). The mixture was filtered, and the collected solids were washed with water and dried, yielding 405 g.
The 405 g were combined with a second batch of comparable quality (152 g). 2 L of ethyl acetate and 6 L of water were added, followed by 480 ml_ of aqueous sodium hydroxide solution (45 w%). The mixture was stirred at r.t. for 30 min.. The phases were separated. The pH of the aqueous phase was adjusted to pH 2 with ap. 770 ml_ of aqueous hydrochloric acid (37 w%). The mixture was filtered, and the collected solids washed with water and dried to yield 535 g.
1H-NMR (400 MHz, de-DMSO): δ = 7.46 (bs, 2H); 8.66 (s, 2H), 12.72 (bs, 1 H).
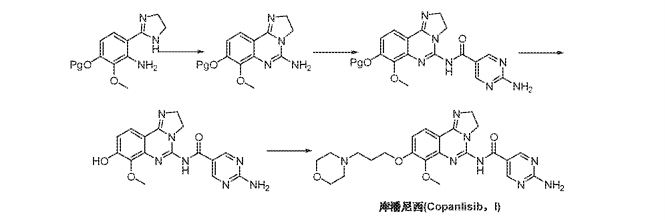
Example 10 : Step A9 : preparation of copanlisib (10)
A mixture of 1250 g of 7-methoxy-8-[3-(morpholin-4-yl)propoxy]-2,3-dihydro-imidazo[1 ,2-c]quinazolin-5-amine, 20.3 kg of N,N-dimethylformamide, 531 g of 2-aminopyrimidine-5-carboxylic acid, 425 g of Ν,Ν-dimethylaminopyridine and 1000 g of N-[3-(dimethylamino)propyl]-N’-ethylcarbodiimide hydrochloride was stirred at r.t. for 17 h. The reaction mixture was filtered. The collected solids were washed with Ν,Ν-dimethylformamide, then ethanol, and dried at 50°C to yield 1 .6 kg (96%). The isolated material was directly converted into the dihydrochloride.
Example 11 : Step A11 : preparation of copanlisib dihydrochloride (11)
To a mixture of 1 .6 kg of copanlisib and 4.8 kg of water were added 684 g of aqueous hydrochloric acid (32 w%) while maintaining the temperature between 20 to 25°C until a pH of 3 to 4 was reached. The resulting mixture was stirred for 10 min, and the pH was checked (pH 3.5). The mixture was filtered, and the filter cake was washed with 0.36 kg of water. 109 g of aqueous hydrochloric acid were added to the filtrate until the pH was 1 .8 to 2.0. The mixture was stirred for 30 min and the pH was checked (pH 1 .9). 7.6 kg of ethanol were slowly added within 5 h at 20 to 25°C, dosing was paused after 20 min for 1 h when crystallization started. After completed addition of ethanol the resulting suspension was stirred for 1 h. The suspension was filtered. The collected solids was washed with ethanol-water mixtures and finally ethanol, and then dried in vacuum to give 1 .57 kg of copansilib dihydrochloride (85 %).
1H-NMR (400 MHz, de-DMSO): δ = 2.32 (m, 2H), 3.1 1 (m, 2H), 3.29 (m, 2H),
3.47 (m, 2H), 3.84 (m, 2H), 3.96 (m, 2H), 4.01 (s, 3H), 4.19 (t, 2H), 4.37 (t, 2H),
4.48 (t, 2H), 7.40 (d, 1 H), 7.53 (bs, 2H), 8.26 (d, 1 H), 8.97 (s, 2H), 1 1 .28 (bs, 1 H), 12.75 (bs, 1 H), 13.41 (bs, 1 H).
HPLC: stationary phase: Kinetex C18 (150 mm, 3.0 mm ID, 2.6 μιτι particle size): mobile phase A: 2.0 ml_ trifluoro acetic acid / 1 L water; mobile phase B: 2.0 ml_ trifluoro acetic acid / L acetonitrile; UV detection at 254 nm switch after 1 minute to 282 nm; oven temperature: 60°C; injection volume: 2.0 μΙ_; flow 1 .7 mL/min; linear gradient after 1 minute isocratic run in 2 steps: 0% B -> 18% B (9 min), 18 % B -> 80% B (2.5 min), 2.5 minutes holding time at 80% B; purity: >99.8% (Rt=6.1 min), relevant potential by-products: 2-Aminopyrimidine-5-carboxylic acid at RRT (relative retention time) of 0.10 (0.6 min) typically <0.01 %, 4-dimethylaminopyrimidine RRT 0.26 (1 .6 min): typically <0.01 %, 7-methoxy-8-[3-(morpholin-4-yl)propoxy]-2,3-dihydroimidazo[1 ,2-c]quinazolin-5-amine RRT 0.40 (2.4 min): typically <0.03 %, by-product 1 RRT 0.93 (5.7 min): typically <0.05 %, by-product 6 RRT 1 .04 (6.4 min): typically <0.05 %, 2-amino- N-{3-(2-aminoethyl)-8-methoxy-7-[3-(morpholin-4-yl)propoxy]-4-oxo-3,4-dihydroquinazolin-2-yl}pyrimidine-5-carboxamicle RRT 1.12 (8.9 min); typically <0.10 %, 5-{[(2-aminopyrimidin-5-yl)carbonyl]amino}-7-methoxy-2,3-dihydroimidazo[ ,2-c]quinazolin-8-yl 2-aminopyrimidine-5-carboxylate RRT 1.41 (8.6 min): typically <0.01 %
Example 15 : Step A11 : further example of preparation of copanlisib dihydrochloride (11)
7.3 g of hydrochloric acid were added to a mixture of 12 g of copanlisib and 33 g of water at maximum 30°C. The resulting mixture was stirred at 25°C for 15 min, and the filtered. The filter residue was washed with 6 g of water. 1 1 .5 g of ethanol were added to the filtrate at 23°C within 1 hour. After the addition was completed the mixture was stirred for 1 hour at 23°C. Additional 59 g of ethanol were added to the mixture with 3 hours. After the addition was completed the mixture was stirred at 23°C for 1 hour. The resulting suspension was filtered. The collected crystals were washed three times with a mixture of 1 1 .9 g of ethanol and 5.0 g of water and the air dried to give 14.2 g of copanlisib dihydrochloride as hydrate I.
Purity by HPLC: > 99.8%; < 0.05% 2-amino-N-{3-(2-aminoethyl)-8-methoxy-7- [3-(morpholin-4-yl)propoxy]-4-oxo-3,4-dihydroquinazolin-2-yl}pyrimidine-5-carboxamide
Example 16 : Step A11 : further example of preparation of copanlisib dihydrochloride (11 )
9.1 kg of hydrochloric acid (25 w%) were added to a mixture of 14,7 kg of copanlisib and 41.9 kg of water at maximum temperature of 28°C. The resulting mixture was stirred at 23°C for 80 minutes until a clear solution was formed. The solution was transferred to a second reaction vessel, and the transfer lines rinsed with 6 kg of water, 14.1 kg of ethanol were slowly added within 70 minutes at 23°C. After the addition of ethanol was completed the mixture was stirred at 23°C for 1 hour. Additional 72.3 kg of ethanol were slowly added within 3.5 hours at 23°C, and resulting mixture stirred at this temperature for 1 hour. The suspension is filtered, and the collected solids were washed twice with 31 kg of an ethanol-water mixture (2.4: 1 (w w)). The product was dried in vacuum with a maximum jacket temperature of 40°C for 3.5 hours to yield 15.0 kg of copanlisib dihydrochloride as hydrate I.
Purity by HPLC: > 99.9 %; < 0.05% 2-amino-N-{3-(2-aminoethyl)-8-methoxy-7-[3-(morpholin-4-yl)propoxy]^-oxo-3,4-dihydroquinazolin-2-yl}pyrimidine-5-carboxamideLoss on drying: 14.7 w%
PATENT
PAPER
ChemMedChem (2016), 11(14), 1517-1530.
2-Amino-N-{7-methoxy-8-[3-(morpholin-4-yl)propoxy]-2,3-dihydroimidazo[1,2-c]quinazolin-5-yl}pyrimidine-5-carboxamide (BAY 80-6946, 39i):
Amine 36 (80% purity; 100 mg, 0.22 mmol) was dissolved in DMF (5 mL), and acid 39i’ (46 mg, 0.33 mmol) was added. PyBOP (173 mg, 0.33 mmol) and DIPEA (0.16 mL, 0.89 mmol) were sequentially added, and the mixture was stirred at RT overnight. EtOAc was added, and the solids were isolated by vacuum filtration to give 39i (42.7 mg, 40%):
1H NMR ([D6 ]DMSO+ 2 drops [D]TFA): d=2.25 (m, 2H), 3.18 (m, 2H), 3.31 (m, 2H), 3.52 (m, 2H), 3.65 (brt, 2H), 4.00 (s, 3H), 4.04 (m, 2H), 4.23 (m, 2H), 4.34 (brt, 2H), 4.54 (m, 2H), 7.43 (d, 1H), 8.04 (d, 1H), 9.01 (s, 2H);
1H NMR of the bis-HCl salt (500 MHz, [D6 ]DMSO): d=2.30–2.37 (m, 2H), 3.11 (brs, 2H), 3.25–3.31 (m, 2H), 3.48 (d, J=12.1 Hz, 2H), 3.83–3.90 (m, 2H), 3.95–4.00 (m, 2H), 4.01 (s, 3H), 4.17–4.22 (m, 2H), 4.37 (t, J=6.0 Hz, 2H), 4.47 (t, J=9.7 Hz, 2H), 7.40 (d, J= 9.2 Hz, 1H), 7.54 (s, 2H), 8.32 (d, J=9.2 Hz, 1H), 8.96 (s, 2H), 11.46 (brs, 1H), 12.92 (brs, 1H), 13.41 (brs, 1H);
13C NMR (125 MHz, [D6 ]DMSO): d=23.09, 45.22, 46.00, 51.21, 53.38, 61.54, 63.40, 67.09, 101.18, 112.55, 118.51, 123.96, 132.88, 134.35, 148.96, 157.25, 160.56, 164.96, 176.02 ppm;
MS (ESI+) m/z: 481 [M+H]+ .
References
- Jump up^ “Phase II Data of Bayer’s Novel Cancer Drug Candidate Copanlisib to be Presented”. Retrieved 3 March 2015.
- Jump up^ Loguidice, Christina (8 December 2014). “Copanlisib Continues to Show Promise for Treating Indolent Lymphomas”. Rare Disease Report. Retrieved 3 March 2015.
- Jump up^ HealthCare, Bayer. “Bayer Advances Clinical Development Program for Investigational Cancer Drug Copanlisib”. www.prnewswire.com.
- Jump up^ “Copanlisib in Treating Patients With Persistent or Recurrent Endometrial Cancer – Full Text View – ClinicalTrials.gov”.
- Jump up^ “Phase II Copanlisib in Relapsed/Refractory Diffuse Large B-cell Lymphoma (DLBCL) – Full Text View – ClinicalTrials.gov”.
- Jump up^ “Copanlisib (BAY 80-6946) in Combination With Gemcitabine and Cisplatin in Advanced Cholangiocarcinoma – Full Text View – ClinicalTrials.gov”.
- Jump up^ “Open-label, Uncontrolled Phase II Trial of Intravenous PI3K Inhibitor BAY80-6946 in Patients With Relapsed, Indolent or Aggressive Non-Hodgkin’s Lymphomas – Full Text View – ClinicalTrials.gov”.
- Jump up^ “Study of Copanlisib in Combination With Standard Immunochemotherapy in Relapsed Indolent Non-Hodgkin’s Lymphoma (iNHL) – Full Text View – ClinicalTrials.gov”.
- Jump up^ “Copanlisib and Rituximab in Relapsed Indolent B-cell Non-Hodgkin’s Lymphoma (iNHL) – Full Text View – ClinicalTrials.gov”.
- Jump up^ “Phase III Copanlisib in Rituximab-refractory iNHL – Full Text View – ClinicalTrials.gov”.
| WO2016071426A1 * | 5 Nov 2015 | 12 May 2016 | Bayer Pharma Aktiengesellschaft | Synthesis of copanlisib and its dihydrochloride salt |
| CN1688582A * | 18 Sep 2003 | 26 Oct 2005 | 拜尔药品公司 | 稠合吡咯-嘧啶衍生物 |
| CN101631464A * | 5 Dec 2007 | 20 Jan 2010 | 拜耳先灵医药股份有限公司 | 用于治疗过度增殖疾病和血管发生相关性疾病的2,3-二氢咪唑并[1,2-c]喹唑啉取代衍生物 |
| CN105130997A * | 25 Sep 2015 | 9 Dec 2015 | 苏州明锐医药科技有限公司 | 一种库潘尼西的制备方法 |
| CN105130998A * | 25 Sep 2015 | 9 Dec 2015 | 苏州立新制药有限公司 | 库潘尼西的制备方法 |
FDA Orange Book Patents
| FDA Orange Book Patents: 1 of 3 | |
|---|---|
| Patent |
US 7511041 |
| Expiration | May 13, 2024 |
| Applicant | BAYER HEALTHCARE |
| Drug Application | N209936 (Prescription Drug: ALIQOPA. Ingredients: COPANLISIBDIHYDROCHLORIDE) |
 |
|
| Names | |
|---|---|
| IUPAC name
2-Amino-N-[7-methoxy-8-(3-morpholin-4-ylpropoxy)-2,3-dihydroimidazo[1,2-c]quinazolin-5-yl]pyrimidine-5-carboxamide
|
|
| Other names
BAY 80-6946
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
| KEGG | |
| MeSH | 2-amino-N-(7-methoxy-8-(3-morpholinopropoxy)-2,3-dihydroimidazo(1,2-c)quinazolin-4-yl)pyrimidine-5-carboxamide |
| UNII | |
| Properties | |
| C23H28N8O4 | |
| Molar mass | 480.53 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
////////////copanlisib, BAY 80-6946, BAYER, orphan drug status, follicular lymphoma, FDA 2017, BAY 84-1236
COC1=C(C=CC2=C1N=C(N3C2=NCC3)NC(=O)C4=CN=C(N=C4)N)OCCCN5CCOCC5