














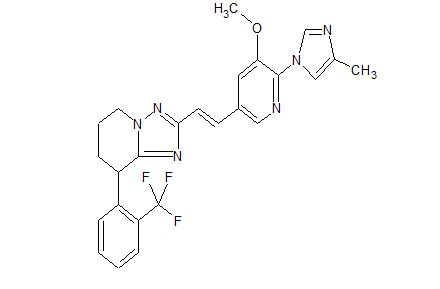
C25 H23 F3 N6 O, 480.48
CAS 1123197-68-1
(+) -2-{(E)-2-[5-methoxy-6-(4-methyl-1H-imidazol-1-yl)pyridin-3-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[1,5-a]pyridine
- (+)-5,6,7,8-Tetrahydro-2-[(1E)-2-[5-methoxy-6-(4-methyl-1H-imidazol-1-yl)-3-pyridinyl]ethenyl]-8-[2-(trifluoromethyl)phenyl][1,2,4]triazolo[1,5-a]pyridine
- (+)-2-[(E)-2-[5-Methoxy-6-(4-methyl-1H-imidazol-1-yl)pyridin-3-yl]ethenyl]-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[1,5-a]pyridine

E2212
CAS 1123197-82-9
- C25 H23 F3 N6 O . 3/2 C4 H6 O6
- [1,2,4]Triazolo[1,5-a]pyridine, 5,6,7,8-tetrahydro-2-[(1E)-2-[6-methoxy-5-(4-methyl-1H-imidazol-1-yl)-2-pyridinyl]ethenyl]-8-[2-(trifluoromethyl)phenyl]-, (8S)-, (2S,3S)-2,3-dihydroxybutanedioate (2:3)
PATENT
https://patents.google.com/patent/US9453000B2/en
Examples 394 and 395 Synthesis of (+) and (−)-2-{(E)-2-[5-methoxy-6-(4-methyl-1H-imidazol-1-yl)pyridin-3-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[1,5-a]pyridine

230 mg of the racemic title compound was obtained from 1-amino-3-(2-trifluoromethylphenyl)piperidin-2-one (343 mg) and (E)-3-[5-methoxy-6-(4-methyl-1H-imidazol-1-yl)pyridin-3-yl]acrylic acid (500 mg) by the same method as in Examples 194 and 195. The racemic title compound (220 mg) was separated by CHIRALPAK™ IC manufactured by Daicel Chemical Industries, Ltd. (2 cm×25 cm; mobile phase: methanol) to obtain the title optically active compound with positive optical rotation and a retention time of 16 minutes (92 mg) and the title optically active compound with negative optical rotation and a retention time of 19 minutes (79 mg).
The property value of the title optically active compound with a retention time of 16 minutes is as follows.
ESI-MS; m/z 481 [M++H].
The property values of the title optically active compound with a retention time of 19 minutes are as follows.
ESI-MS; m/z 481 [M++H]. 1H-NMR (CDCl3) δ (ppm): 1.90-2.01 (m, 1H), 2.10-2.35 (m, 2H), 2.29 (s, 3H), 2.43-2.52 (m, 1H), 3.95 (s, 3H), 4.27-4.41 (m, 2H), 4.69 (dd, J=6.0, 8.4 Hz, 1H), 7.02 (d, J=8.0 Hz, 1H), 7.08 (d, J=16.4 Hz, 1H), 7.40 (dd, J=7.6, 7.6 Hz, 1H), 7.44-7.53 (m, 4H), 7.73 (d, J=8.0 Hz, 1H), 8.13 (d, J=1.6 Hz, 1H), 8.34 (s, 1H).
PATENT
WO2009028588
https://patentscope.wipo.int/search/en/detail.jsf;jsessionid=D6AD22B6CC7302560AE1ADCED305CDCE.wapp2nC?docId=WO2009028588&tab=FULLTEXT&queryString=%28PA%2Feisai%29%2520&recNum=93&maxRec=725
(+)および(-)-2-{(E)-2-[5-メトキシ-6-(4-メチル-1H-イミダゾール-1-イル)ピリジン-3-イル]ビニル}-8-(2-トリフルオロメチルフェニル)-5,6,7,8-テトラヒドロ-[1,2,4]トリアゾロ[1,5-a]ピリジンの合成
[化221]
実施例194および実施例195と同様の方法により、1-アミノ-3-(2-トリフルオロメチルフェニル)ピペリジン-2-オン(343mg)および(E)-3-[5-メトキシ-6-(4-メチル-1H-イミダゾール-1-イル)ピリジン-3-イル]アクリル酸(500mg)から、ラセミ体の表題化合物を230mg得た。ラセミ体の表題化合物(220mg)をダイセル製CHIRALPAK TM IC(2cm×25cm:移動相;メタノール)にて分取し、(+)の旋光性を有する保持時間16分の表題光学活性化合物(92mg)および(-)の旋光性を有する保持時間19分の表題光学活性化合物(79mg)を得た。
保持時間16分の表題光学活性体の物性値は以下の通りである。
ESI-MS;m/z 481[M ++H].
保持時間19分の表題光学活性体の物性値は以下の通りである。
ESI-MS;m/z 481[M ++H]. 1H-NMR(CDCl 3)δ(ppm):1.90-2.01(m,1H),2.10-2.35(m,2H),2.29(s,3H),2.43-2.52(m,1H),3.95(s,3H),4.27-4.41(m,2H),4.69(dd,J=6.0,8.4Hz,1H),7.02(d,J=8.0Hz,1H),7.08(d,J=16.4Hz,1H),7.40(dd,J=7.6,7.6Hz,1H),7.44-7.53(m,4H),7.73(d,J=8.0Hz,1H),8.13(d,J=1.6Hz,1H),8.34(s,1H).
Example 394 and Example 395
(+) and (−)-2-{(E) -2- [5-methoxy-6- (4-methyl-1H-imidazol-1-yl) pyridin-3-yl] Synthesis of vinyl} -8- (2-trifluoromethylphenyl) -5,6,7,8-tetrahydro- [1,2,4] triazolo [1,5-a] pyridine [Formula
221]
Example 194 and By a method similar to Example 195, 1-amino-3- (2-trifluoromethylphenyl) piperidin-2-one (343 mg) and (E) -3- [5-methoxy-6- (4-methyl-) 1 H-Imidazol-1-yl) pyridin-3-yl] acrylic acid (500 mg) gave 230 mg of the racemic title compound. Racemic title compound (220 mg) a Daicel CHIRALPAK TM IC (2 cm × 25 cm: mobile phase; methanol) was collected by min (+) title optically active compound of the retention time of 16 minutes with a optical rotation of (92 mg) The title optically active compound (79 mg) having a polarizability of (−) and a retention time of 19 minutes was obtained.
The physical property values of the title optically active substance with a retention time of 16 minutes are as follows.
ESI-MS; m / z 481 [M + + H].
The physical property values of the title optically active substance with a retention time of 19 minutes are as follows.
ESI-MS; m / z 481 [M + + H]. 1 H-NMR (CDCl 3)) Δ (ppm): 1.90 to 2.01 (m, 1 H), 2.10 to 2.35 (m, 2 H), 2.29 (s, 3 H), 2.43 to 2.52 (m) , 1 H), 3.95 (s, 3 H), 4.27-4. 41 (m, 2 H), 4.69 (dd, J = 6.0, 8.4 Hz, 1 H), 7.02 (d , J = 8.0 Hz, 1 H), 7.08 (d, J = 16.4 Hz, 1 H), 7.40 (dd, J = 7.6, 7.6 Hz, 1 H), 7.44-7. 53 (m, 4H), 7.73 (d, J = 8.0 Hz, 1 H), 8.13 (d, J = 1.6 Hz, 1 H), 8.34 (s, 1 H).
PATENT
https://patents.google.com/patent/WO2010098490A1/it

As a novel compound that has an effect of reducing the production of Aβ40 and
42 and is expected as a therapeutic or prophylactic agent for Alzheimer’s disease or the like, the present inventors have found a compound represented by the following formula (1) (compound
(D): [Formula 1]
and filed a patent application for the invention (PCT/JP08/065365).
Generally, properties of salts of compounds and those crystals that are useful as pharmaceuticals are highly important for the development of pharmaceuticals, because the properties greatly affect bioavailability of drugs, purity of drug substances, formulation of preparations, and the like. Therefore, it is necessary to research which salts and crystal forms of the compound of the formula (1) are most excellent as pharmaceuticals. Specifically, since their properties depend on the character of the individual compounds, it is generally difficult to estimate salts and crystal forms for drug substances having excellent properties and it is demanded to actually make various studies for each compound.
EXAMPLES [0023] The present invention will be described in detail below with reference to reference examples and examples; however, the present invention is not limited to these reference examples and examples. [0024]
The following abbreviations are used in the following reference examples and examples.
DMF: N,N’-dimethylformamide
THF: Tetrahydrofuran
EDC: lrEmyl-S-β-dimemylammopropytycarbodiimide hydrochloride HOBT: 1-Hydroxybenzotriazole IPEA: Diisopropylethylamine [0025]
In powder X-ray diffractometry of the crystals produced in the following examples, the resulting crystals were placed on a sample stage of a powder X-ray diffractometer and analyzed under the following conditions. [0026] Measurement conditions
Sample holder: Aluminum Target: Copper
Detector: Scintillation counter Tube voltage: 50 kV Tube current: 300 mA
Slit: DS 0.5 mm (Height limiting slit 2 mm), SS Open, RS Open Scanning rate : 5 °/min
Sampling interval: 0.02° Scan range: 5 to 35° Goniometer: Horizontal goniometer [0027] Reference Example 1
Svnmesis ofr8SV2-(fE)-246-memoxy-5-(4-memyl-lH-imidazol-l-vnpyridin-2-yllvmvU-8-(2-trifluoromethylphenyl‘)-5,6J,8-tetrahvdro-[1.2,41triazolo[l.,5-a]pyridine
[Formula 2]
Synthesis of l-amino-3-(2-trifluoromemylphenyl)piperidin-2-one Thionyl chloride (2.72 mL) was added to a solution of 2-trifluoromethylphenylacetic acid (1.9 g) in methanol (38 mL), followed by stirring at room temperature for three hours. The reaction solution was concentrated under reduced pressure. The resulting residue was diluted with DMF. Sodium hydride (containing 40% mineral oil, 410 mg) was added under ice-cooling, followed by stirring for 10 minutes. The reaction solution was further stirred for 30 minutes and then ice-cooled again. l-Chloro-3-iodopropane (1.02 mL) was added to the reaction mixture, and the reaction solution was stirred at room temperature overnight. Water and ethyl acetate were added to the reaction mixture, and the organic layer was separated. The resulting organic layer was washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was diluted with ethanol (26.6 mL). Hydrazine monohydrate (7.6 mL) was added, and the reaction solution was stirred at room temperature for two hours and then at 60°C for further three hours. The reaction mixture was concentrated under reduced pressure. Saturated aqueous sodium bicarbonate and ethyl acetate and were added to the residue, and the organic layer was separated. The resulting organic layer was washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (carrier: Chromatorex NH; elution solvent: heptane-ethyl acetate system) to obtain 1.68 g of the title compound. The property values of the compound are as follows.
ESI-MS; m/z 259 [M+H-H]. 1H-NMR (CDCl3) δ (ppm): 1.82-2.10 (m, 3H), 2.18-2.26 (m, IH), 3.58-3.76 (m, 2H), 4.07 (dd, J = 10.0, 5.6 Hz, IH), 4.60 (s, 2H), 7.24 (d, J = 7.6 Hz, IH), 7.35 (t, J = 7.6 Hz, IH), 7.51 (t, J = 7.6 Hz, IH)5 7.66 (d, J = 7.6 Hz, IH). [0028] Synthesis of (EV3-[6-methoxy-5-(4-methyl- 1 H-imidazol- 1 -yl)pyridin-2-yl]-N-f2-oxo-3 -(2-trifluoromethylphenyl)piperidin- 1 -yl]acrylamide
EDC (834 mg), HOBT (588 mg) and IPEA (2.03 mL) were added to a suspension of (E)-3-[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridm-2-yl]acrylic acid trifluoroacetate (1.42 g) and l-amήio-3-(2-trifluoromethylphenyl)piperidin-2-one (750 mg) in DMF (30 mL). After stirring at room temperature for 14 hours, a saturated sodium bicarbonate solution and ethyl acetate were added to the reaction solution, and the organic layer was separated. The resulting organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (carrier: Chromatorex NH; elution solvent: ethyl acetate-methanol system) to obtain 1.23 g of the title compound. The property values of the compound are as follows. ESI-MS; m/z 500 [M1H-HJ. [0029]
Synthesis of r8S>-2-(fEV2-r6-methoxy-5-r4-methyl-lH-imidazol-l-vnpyridm’2-vnvinvU-8-(2-trifluoromethvlphenvD-5.6.7.8-tetrahvdro-ri.2.41triazoloπ.5-a1pvridine Phosphorus oxychloride (24.2 mL) was added to (E)-3~[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]-N-[2-oxo-3-(2-trifluoromethylphenyl)piperidin-l-yl]acrylamide (1.2 g). The reaction solution was stirred at 1000C for one hour and then concentrated under reduced pressure. Subsequently, the residue was diluted with acetic acid (24.2 mL) and then ammonium acetate (1.9 g) was added, followed by stirring at 1500C for two hours. The reaction solution was left to cool to room temperature and then concentrated under reduced pressure. A saturated sodium bicarbonate solution and ethyl acetate were added to the resulting residue, and the organic layer was separated. The resulting organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (carrier: Chromatorex NH; elution solvent: heptane-ethyl acetate system) to obtain a racemate of the title compound (750 mg). The resulting racemate (410 mg) was separated by CHIRALP AK™ IA manufactured by Daicel Chemical Industries, Ltd. (2 cm x 25 cm, mobile phase: hexane:ethanol = 8:2, flow rate: 10 mL/min) to obtain the title compound with a retention time of 33 minutes and negative optical rotation (170 mg) as crystals. The property values of the title compound are as follows.
1H-NMR (CDCl3) δ (ppm): 1.90-2.01 (m, IH), 2.10-2.35 (m, 2H), 2.29 (d, J = 1.2 Hz, 3H), 2.42-2.51 (m, IH), 4.03 (s, 3H), 4.28-4.41 (m, 2H), 4.70 (dd, J = 8.4, 6.0 Hz, IH), 6.92 (d, J = 8.0 Hz, IH), 6.95 (t, J = 1.2 Hz, IH), 7.01 (d, J = 7.6 Hz, IH), 7.39 (t, J = 7.6 Hz5 IH), 7.44 (d, J = 16.0 Hz, IH), 7.45 (d, J = 8.0 Hz, IH), 7.49 (t, J = 7.6 Hz, IH), 7.63 (d, J = 16.0 Hz5 IH), 7.72 (d, J = 7.6 Hz, IH), 7.76 (d, J = 1.2 Hz, IH). [0030]
(8S)-2-{(E)-2-[6-Methoxy-5-(4-methyl-lH-imidazol-l-yl)ρyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahydro-[l,2,4]triazolo[l,5-a]pyridine synthesized according to the above reference example was used for the following synthesis of salts. [0031] Example 1
Synthesis of r8SV2-{rEV2-[6-methoxy-5-(4-methyl-lH-imidazol-l-vπpyridin-2-vnvinvU-8-f2-trifluoromethylphenyl)-5.6.7.8-tetrahvdro-fl,2,4]triazolo[l.,5-a]pyridine 1.5 D-tartrate
(8S)-2-{(E)-2-[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahydro-[l,2,4]triazolo[l,5-a]pyridine (33.70 mg) was dissolved in 285 μL of a D-tartaric acid-ethanol solution (110.92 mg/3 mL) with stirring at room temperature. The oil was precipitated when 1 mL of heptane was added. Accordingly, the oily substance was dissolved by adding 1 mL of ethanol. Further, 0.5 mL of heptane was added, and the mixture was transferred to a low temperature laboratory at about 50C (under shading) and continuously stirred for 24 hours. Thus, partial gelation occurred. Thereafter, the mixture was brought back to room temperature and continuously stirred, resulting in precipitation of a solid. The solid was collected by filtration through a glass filter and dried under reduced pressure at room temperature to obtain 21.25 mg of the title compound as white solid crystals. 1H-NMR (600 MHz, DMSOd6) δ (ppm): 1.96 (m, IH), 2.14 (s, 3H), 2.16 (m, 2H), 2.29 (m, IH), 3.98 (s, 3H), 4.28 (m, 2H), 4.29 (s, 3H), 4.51 (dd, J = 9, 6 Hz, IH), 7.22 (s, IH), 7.25 (brd, J = 8 Hz, IH), 7.27 (d, J = 8 Hz, IH), 7.32 (d, J = 16 Hz, IH)5 7.46 (d, J = 16 Hz, IH), 7.49 (brdd, J = 8 Hz, IH), 7.61 (brdd, J = 8 Hz5 IH), 7.77 (brd, J = 8 Hz, IH), 7.78 (d, J = 8 Hz, IH), 7.91 (s, IH). [0032] Example 2
Synthesis of (‘8SV2-l(Ε)-2-f6-methoxy-5-(4-methyl-lH-imidazol-l-vnpyridm-2-yllvinyl>-8-f2-trifluoromethylphenylV5,6J,8-tetrahvdro-[l ,2,4]triazolo[l ,5-a]pyridine di-D-tartrate
(8S)-2-{(E)-2-[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,657,8-tetrahydro-[l ,2,4]triazolo[l ,5-a]ρyridine (810.18 mg) was dissolved in 8 mL of a D-tartaric acid-ethanol solution (751.13 mg/10 mL) with stirring at room temperature. The oil was precipitated when 2 mL of heptane was added. Accordingly, the oily substance was dissolved by ultrasonic treatment to prepare a clear solution. Several mg of crystals of the 1.5 D-tartrate prepared according to Example 1 were added, followed by stirring at room temperature. Stirring for about one hour resulted in gelation and subsequent precipitation of a solid. Further, stirring was continued while gradually adding 14 mL of heptane. A part of the suspension (2 mL) was separated and the solid was collected by filtration through a glass filter. The solid was dried under reduced pressure at room temperature to obtain 71.14 mg of the title compound as white solid crystals. 1H-NMR (400 MHz, DMSOd6) δ (ppm): 1.97 (m, IH), 2.15 (s, 3H), 2.16 (m, 2H), 2.30 (m, IH), 3.98 (s, 3H), 4.28 (m, 2H), 4.29 (s, 4H), 4.51 (dd, J = 9, 6 Hz, IH), 7.22 (brs, IH), 7.25 (brd, J = 8 Hz, IH), 7.27 (d, J = 8 Hz, IH), 7.32 (d, J = 16 Hz, IH), 7.46 (d, J = 16 Hz, IH), 7.49 (brdd, J – 8 Hz, IH), 7.61 (brdd, J = 8 Hz, IH), 7.77 (brd, J = 8 Hz, IH), 7.78 (d, J = 8 Hz, IH), 7.91 (brs, IH). [0033] Example 3
Synthesis of r8SV2-(rE)-2-r6-methoxy-5-r4-methyl-lH-imidazol-l-vnpyridin-2-yl1vinvU-8-α-trifluoromethylphenyl)-5,6J,8-tetrahydro-[1.2,4]triazolo[l,5-a]pyridine disulfate
Concentrated sulfuric acid (11.5 μL) was added to a solution of (8S)-2-{(E)-2-[6- methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-txifluoromethylphenyl)-5,6,7,8-tetrahydro-[l52,4]triazolo[l55-a]pyridine (98.09 mg) in ethanol (1 mL), and 1 mL of ethyl acetate was added with stirring at room temperature. Since the oily portion was confirmed on the bottom of the recovery flask, the oily substance was dissolved by ultrasonic treatment. Stirring at room temperature under shading for about 30 minutes resulted in precipitation of a solid. The solid was collected by filtration through a glass filter and dried under reduced pressure at room temperature to obtain 127.94 mg of the title compound as white solid crystals. 1H-NMR (400 MHz, DMSOd6) δ (ppm): 1.97 (m, IH), 2.17 (m, 2H), 2.30 (m, IH), 2.34 (brd, J = 1 Hz, 3H), 4.01 (s, 3H), 4.29 (m, 2H), 4.52 (dd, J = 9, 6 Hz, IH)5 7.25 (brd, J = 8 Hz, IH), 7.37 (d, J = 16 Hz, IH), 7.40 (d, J = 8 Hz, IH), 7.50 (brdd, J = 8 Hz, IH), 7.55 (d, J = 16 Hz, IH), 7.61 (brdd, J = 8 Hz, IH), 7.77 (m, IH), 7.78 (m, IH), 8.00 (d, J = 8 Hz, IH), 9.36 (d, J = 2 Hz, IH). [0034] Example 4 Synthesis of (8SV2-((E)-2-[“6-methoxy-5-(4-methyl-lH-imidazol-l-ylN)ρyridin-2-yllvinvU-8-(‘2-trifluoromethylphenyl)-5,6,7,8-tetrahvdiO-[1.2,41triazolo[l,5-a]pyridine dihydrobromide
Concentrated hydrobromic acid (24.8 μL) was added to a solution of (8S)-2-{(E)-2-[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,6,7,84etrahydro-[l,254]triazolo[l55-a]pyridine (51.42 mg) m ethanol (1 mL), and 1 mL of heptane was added with stirring at room temperature. After several minutes, 1 mL of heptane was further added to the solution and stirring was continued. The solution was stirred at room temperature for one hour and then further stirred at about 50C for 20 minutes. The precipitated solid was collected by filtration through a glass filter and dried under reduced pressure at room temperature to obtain 49.24 mg of the title compound as white solid crystals. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 1.99 (m, IH), 2.17 (m, 2H), 2.30 (m, IH), 2.34 (brd, J = 1 Hz5 3H), 4.01 (s, 3H), 4.30 (m, 2H), 4.52 (dd, J = 9, 6 Hz5 IH), 7.25 (brd, J = 8 Hz5 IH), 7.37 (d, J = 16 Hz, IH), 7.40 (d, J = 7 Hz, IH)57.50 (brdd, J = 8 Hz, IH), 7.55 (d, J = 16 Hz, IH), 7.61 (brdd, J = 8 Hz5 IH), 7.77 (m, IH)5 7.78 (m, IH), 8.00 (d, J = 7 Hz, IH), 9.37 (d, J = 2 Hz, IH). [0035] Example 5
Synthesis of r8SV2-((Ε)-2-r6-methoxy-5-r4-methyl-lH-imidazol-l-yl)ρyridin-2-vnvinyl}-8-r2-trifluoromethylphenyl)-5,6J,8-tetrahvdro-[1.2,41triazolo[1.5-alpyridine hydrochloride
Concentrated hydrochloric acid (3.6 μL) was added to a solution of (8S)-2-{(E)- 2-[6-methoxy-5-(4-metiiyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylplienyl)-5,6,7,8-te1xahydro-[l,2,4]triazolo[l,5-a]pyridme (19.80 mg) in 2-propanol (1 mL), and a total of 4 mL of heptane was added in 1 mL portions with stirring at room temperature. The solution was stirred at room temperature under shading for five days. The precipitated solid was collected by filtration through a glass filter and dried under reduced pressure at room temperature to obtain 7.45 mg of the title compound as white solid crystals. 1H-NMR (400 MHz, DMSOd6) δ (ppm): 1.97 (m, IH), 2.17 (m, 2H)5 2.30 (m, IH), 2.30 (s, 3H), 4.00 (s, 3H), 4.30 (m, 2H)5 4.52 (dd, J = 9, 6 Hz5 IH), 7.25 (brd, J – 8 Hz5 IH), 7.36 (d, J = 16 Hz5 IH), 7.37 (d5 J = 8 Hz, IH), 7.50 (brt, J = 8 Hz5 IH)5 7.53 (d, J = 16 Hz5 IH)5 7.61 (brt, J = 8 Hz5 IH)5 7.66 (brs, IH), 7.77 (brd, J = 8 Hz, IH)5 7.96 (d, J = 8 Hz5 IH), 9.06 (brs, IH). [0036] Example 6
Synthesis of (8S)-2-((ΕV2-r6-methoxy-5-(‘4-methyl-lH-imidazol-l-yl)pyridin-2-yl1vinvU-8-(2-trifluoromethylphenyl)-5.6,7,8-tetrahvdro-[l,2,4]triazolo[L5-a1pyridine hydrochloride Concentrated hydrochloric acid (14.3 μL) and heptane (7 mL) were added to a solution of (8S)-2-{(E)-2-[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,657,8-tetrahydro-[l,2,4]triazolo[l,5-a]pyridine (79.77 mg) in 2-propanol (3 mL). A small amount of the crystals obtained in Example 5 were added as seed crystals with stirring at room temperature. The mixture was transferred to a low temperature laboratory at about 50C and stirred for one hour. Thereafter, 1 mL of heptane was further added, followed by stirring for several minutes. When the precipitated solid was collected by filtration through a glass filter, the solid was precipitated in the filtrate. The precipitated solid was collected by filtration through a glass filter and dried under reduced pressure at room temperature to obtain 38.02 mg of the title compound as white solid crystals. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 1.97 (m, IH), 2.17 (m5 2H), 2.29 (m, IH), 2.32 (brd, J = 1 Hz, 3H), 4.00 (s, 3H), 4.30 (m, 2H), 4.52 (dd, J = 9, 6 Hz, IH), 7.25 (brd, J = 8 Hz, IH), 7.37 (d, J = 16 Hz5 IH), 7.38 (d, J = 8 Hz, IH), 7.50 (brdd, J = 8 Hz, IH)5 7.54 (d, J = 16 Hz, IH), 7.61 (brdd, J = 8 Hz, IH), 7.72 (brs, IH), 7.77 (brd, J = 8 Hz, IH), 7.98 (d, J = 8 Hz5 IH)5 9.24 (brs, IH). [0037] Example 7
SvnJhesis off8SV2-f(E>2-r6-memoxy-5-(4-mefovπ trifluoromethylt>henylV5,6,7,8-tetrahvdro-[l,2,4]triazolo[l,5-a]pyridine mesylate
Mesylic acid (0.8 μL) was added to a mixed solution of (8S)-2-{(E)-2-[6- methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylphen^ tetrahydro-[l52,4]triazolo[l,5-a]pyridine (50 mg) in t-butyl methyl ether (0.8 mL)-ethaαol (0.1 mL). The mixture was solidified as a result of stirring at room temperature for two hours. The solid was collected by filtration through a glass filter. The solid was washed with t-butyl methyl ether-ethanol (8:1) and then dried under reduced pressure at room temperature to obtain 51.9 mg of the title compound as pale yellow solid crystals.
1H-NMR (DMSO-d6) δ (ppm): 1.90-2.05 (m, IH)3 2.10-2.22 (m, 2H), 2.28-2.40 (m, IH), 2.31 (s, 3H), 2.35 (s, 3H)5 4.02 (s, 3H)5 4.25-4.39 (m, 2H), 4.50-4.55 (m, IH), 7.27 (d5 J = 8.0 Hz5 IH)5 7.38 (d, J = 16.0 Hz5 IH)5 7.41 (d, J = 8.0 Hz, IH)5 7.51 (t5 J = 8.0 Hz5 IH)5 7.55 (d, J = 16.0 Hz5 IH), 7.63 (t, J = 8.0 Hz5 IH)5 7.78 (d, J = 8.0 Hz5 IH)5 7.79 (s, IH), 8.01 (d, J = 8.0 Hz5 IH), 9.37 (s, IH). [0038] Example 8 Synthesis of (8S)-2-((ΕV2-r6-methoxy-5-(4-methyl-lH-imidazol-l-vnpyridin-2-vnvinvn-8-r2-trifluoromethylphenyl)-5.6,7,8-tetrahydro-[l.,2,4‘|triazolo[l,5-a]pyridine diphosphate
A solution of phosphoric acid (52.8 mg) in acetonitrile (0.2 mL) was added to a solution of (8S)-2-{(E)-2-[6-methoxy-5-(4-methyl-lH-mτidazol-l-yl)ρyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5565758-tetrahydro-[l5254]triazolo[l,5-a]pyridine (100 mg) in acetonitrile (0.8 mL) at room temperature. The precipitated oil was solidified as a result of stirring with spatula. The solid was collected by filtration through a glass filter. The solid was washed with ice-cold acetonitrile, air-dried at room temperature for 10 minutes and then dried under reduced pressure at room temperature to obtain 120 mg of the title compound as white solid crystals. 1H-NMR (DMSO-d6) δ (ppm): 1.90-2.05 (m, IH), 2.11-2.20 (m, 2H), 2.15 (s, 3H), 2.25-2.35 (m, IH), 3.99 (s, 3H)5 4.24-4.39 (m, 2H), 4.50-4.55 (m, IH)5 7.23 (s, IH), 7.26 (d, J = 7.0 Hz, IH), 7.28 (d, J = 8.0 Hz, IH), 7.33 (d, J = 16.0 Hz5 IH), 7.47 (d, J = 16.0 Hz5 IH), 7.51 (t, J = 7.0 Hz, IH), 7.63 (t, J = 7.0 Hz, IH), 7.78 (d, J = 7.0 Hz, IH), 7.79 (d, J = 8.0 Hz, IH), 7.90 (s, IH). [0039] Example 9 Svnmesis of(8SV2-{(E)-2-[6-memoxy-5-(4-methyl-lH-irnidazol-l-yl)pyridin-2-yl1vinvU-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahvdro-[l .2.41triazolo[l .5-a]pyridine diphosphate
A solution of phosphoric acid (13.2 mg) in ethanol (0.05 mL) was added to a mixed solution of (8S)-2-{(E)-2-[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahydro-[l,2!,4]triazolo[l,5-a]pyridme (50 mg) in heptane (0.6 mL)-ethanol (0.15 mL) at room temperature. The reaction solution was stirred at room temperature, and the precipitated solid was collected by filtration through a glass filter. The solid was washed with heptane-ethanol (3:1) and then dried under reduced pressure at room temperature to obtain 37.6 mg of the title compound as white solid crystals. 1H-NMR (DMSOd6) δ (ppm): 1.90-2.05 (m, IH), 2.11-2.20 (m, 2H), 2.15 (s, 3H), 2.25-2.35 (m, IH), 3.99 (s, 3H), 4.24-4.39 (m, 2H), 4.50-4.55 (m, IH), 7.23 (s, IH), 7.26 (d, J = 7.0 Hz, IH), 7.28 (d, J = 8.0 Hz, IH), 7.33 (d, J = 16.0 Hz, IH), 7.47 (d, J = 16.0 Hz, IH), 7.51 (t, J = 7.0 Hz, IH), 7.63 (t, J = 7.0 Hz, IH), 7.78 (d, J = 7.0 Hz, IH), 7.79 (d, J = 8.0 Hz, IH), 7.90 (s, IH).
CLIP
Development of an Efficient Manufacturing Process for E2212 toward Rapid Clinical Introduction
 , Taiju Nakamura‡, Atsushi Kamada†, Takeo Sasaki§, Toshiyuki Uemura§, Yorihisa Hoshino†, Masaaki Matsuda†, Yongbo Hu∥, Daiju Hasegawa§, Kazato Inanaga‡, Nobuaki Sato§, Kazuhiro Yoshizawa‡, George A. Moniz∥, Gordon D. Wilkie∥, Francis G. Fang∥, Yoshihiro Nishikawa‡, and Katsuya Tagami*‡
, Taiju Nakamura‡, Atsushi Kamada†, Takeo Sasaki§, Toshiyuki Uemura§, Yorihisa Hoshino†, Masaaki Matsuda†, Yongbo Hu∥, Daiju Hasegawa§, Kazato Inanaga‡, Nobuaki Sato§, Kazuhiro Yoshizawa‡, George A. Moniz∥, Gordon D. Wilkie∥, Francis G. Fang∥, Yoshihiro Nishikawa‡, and Katsuya Tagami*‡
Process studies of E2212 (1) toward rapid clinical introduction are described. Through comprehensive route-finding studies and optimization of key condensation and cyclization steps, a racemate-based manufacturing route was established and successfully scaled-up to the hundred kilogram scale. For the rapid delivery of a drug substance containing the Z isomer for preclinical safety studies, the successful scale-up of the photoisomerization of an olefin in a flow system is also presented.
https://pubs.acs.org/doi/10.1021/acs.oprd.8b00444
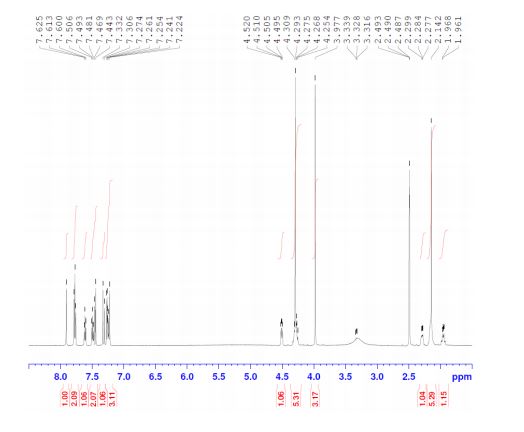
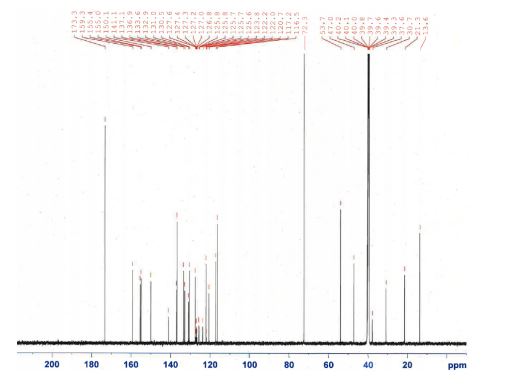
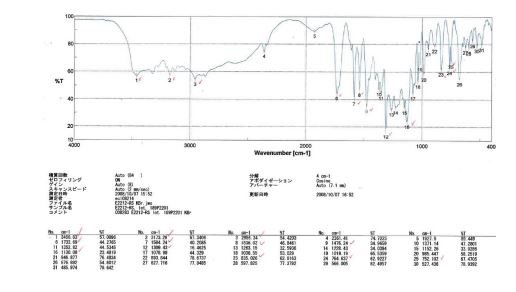
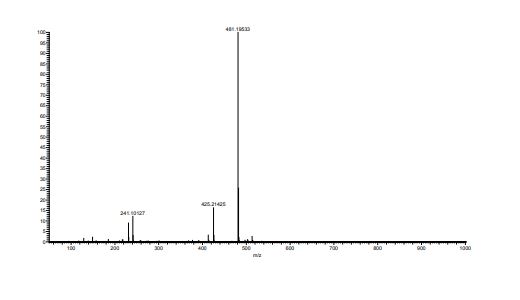
E2212 (1) (18.0 kg, 92.5% yield) as a white solid. Mother liquor 3 were recycled according to the procedure described below. FTIR (cm–1, KBr) 3461, 3173, 2956, 1734, 1584, 1536, 1476, 1309, 1130, 835, 765, 752; 1H NMR (600 MHz, DMSO-d6) δ 7.91 (s, 1H), 7.78 (d, J = 8.4 Hz, 1H), 7.77 (br d, J = 8.4 Hz, 1H), 7.61 (br dd, J = 7.8, 7.8 Hz, 1H), 7.49 (br dd, J = 7.8, 7.8 Hz, 1H), 7.46 (d, J= 15.6 Hz, 1H), 7.32 (d, J = 15.6 Hz, 1H), 7.27 (d, J = 7.8 Hz, 1H), 7.25 (br d, J = 7.8 Hz, 1H), 7.22 (s, 1H), 4.51 (dd, J = 9.0, 6.0 Hz, 1H), 4.29 (s, 3H), 4.28 (m, 2H), 3.98 (s, 3H), 2.29 (m, 1H), 2.14 (s, 3H), 2.16 (m, 2H), 1.96 (m, 1H); 13C NMR (150 MHz, DMSO-d6) δ 173.3, 159.3, 155.4, 155.0, 150.1, 141.1, 137.1, 136.9, 133.6, 132.9, 131.0, 130.5, 127.6, 127.1 (q, JC–F = 30 Hz), 125.8 (q, JC–F = 5.6 Hz), 124.7 (q, JC–F = 270 Hz), 122.2, 120.7, 117.2, 116.5, 72.3, 53.7, 47.0, 37.6, 30.7, 21.3, 13.6; HRMS (ESI+) calcd for C25H23F3N6O ([M + H]+) 481.1958, found 481.1953.
///////////E2212, E 2212