














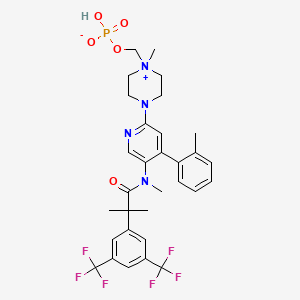

Fosnetupitant
- Molecular FormulaC31H35F6N4O5P
- Average mass688.598 Da
[4-[5-[[2-[3,5-bis(trifluoromethyl)phenyl]-2-methylpropanoyl]-methylamino]-4-(2-methylphenyl)pyridin-2-yl]-1-methylpiperazin-1-ium-1-yl]methyl hydrogen phosphate
CAS 1703748-89-3
HCL 1643757-72-5
FDA 2014 AND EMA 2015
- 07-PNET
In April 2018, the U.S. Food and Drug Administration (FDA) and the Swiss company Helsinn approved the intravenous formulation of AKYNZEO® (NEPA, a fixed antiemetic combination of fosnetupitant, 235mg, and palonosetron, 0.25mg) as an alternative treatment option for patients experiencing chemotherapy-induced nausea and vomiting. Fosnetupitant is the pro-drug form of netupitant. Generally, 25% to 30% of patients with a diagnosis of cancer receive chemotherapy as a treatment modality and 70% to 80% of these patients undergoing chemotherapy treatment may experience nausea and vomiting as major side effects. Considered one of the most distressing side effects of chemotherapy, nausea and vomiting has an immense impact on the quality of life of patients receiving certain antineoplastic therapies.
In April 2018, the U.S. Food and Drug Administration (FDA) and the Swiss company Helsinn approved the intravenous formulation of AKYNZEO® (NEPA, a fixed antiemetic combination of fosnetupitant, 235mg, and palonosetron, 0.25mg) as an alternative treatment option for patients experiencing chemotherapy-induced nausea and vomiting 3. Fosnetupitant is the pro-drug form of netupitant Label.
Generally, 25% to 30% of patients with a diagnosis of cancer receive chemotherapy as a treatment modality and 70% to 80% of these patients undergoing chemotherapy treatment may experience nausea and vomiting as major side effects. Considered one of the most distressing side effects of chemotherapy, nausea and vomiting has an immense impact on the quality of life of patients receiving certain antineoplastic therapies 1.
Fosnetupitant: Fosnetupitant is a selective antagonist of human substance P/neurokinin 1 (NK-1) receptors. Upon intravenous administration, Fosnetupitant is converted by phosphatases to its active form. It competitively binds to and blocks the activity of NK-1 receptors in the central nervous system, by inhibiting binding of substance P (SP) to NK-1 receptors. This prevents delayed emesis, which is associated with SP secretion. AKYNZEO is a combination of palonosetron, a serotonin-3 receptor antagonist, and Fosnetupitant (capsules for oral use) or Fosnetupitant (injections for intravenous use). AKYNZEO for injection is indicated in combination with dexamethasone in adults for the prevention of acute and delayed nausea and vomiting associated with initial and repeat courses of highly emetogenic cancer chemotherapy.
EMA
The chemical name of fosnetupitant chloride hydrochloride is 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)- N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-((phosphonooxy)methyl)piperazin-1- ium chloride hydrochloride is corresponding to the molecular formula C31H37Cl2N4O5P. It has a relative molecular mass of 761.53 g/mol and the following structure:
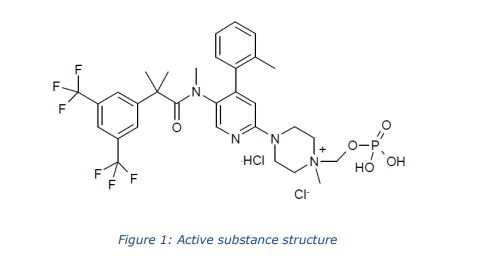
The chemical structure of fosnetupitant chloride hydrochloride was elucidated by a combination of 1H and 13C NMR spectroscopy, infrared spectroscopy, mass spectrometry and elemental analysis. The active substance is achiral. The solid state properties of the active substance were measured by gravimetric vapour sorption and x-ray powder diffraction (XRPD). It is a white to off-white to yellowish, crystalline, hygroscopic solid. Three polymorphic forms have been identified following extensive screening, requiring isolation from different solvent mixtures. Fosnetupitant chloride hydrochloride is always isolated as form I following the commercial manufacturing process. Since it is dissolved and lyophilised during finished product manufacture, particle size and polymorphic form are not considered critical quality attributes (CQAs) of the active substance and are not included in the specification.
RX
AKYNZEO (300 mg netupitant/0.5 mg palonosetron) capsules are an oral combination product of netupitant, a substance P/neurokinin 1 (NK-1) receptor antagonist, and palonosetron hydrochloride, a serotonin-3 (5-HT3) receptor antagonist. Both netupitant and palonosetron hydrochloride are anti-nausea and anti-emetic agents.
Netupitant is chemically described: 2-[3,5-bis(trifluoromethyl)phenyl]-N, 2 dimethyl-N-[4-(2methylphenyl)-6-(4-methylpiperazin-1-yl)pyridin-3-yl] propanamide. The empirical formula is C30H32F6N4O, with a molecular weight of 578.61. Netupitant exists as a single isomer and has the following structural formula:
 |
Palonosetron hydrochloride is chemically described: (3aS)-2-[(S)-1-Azabicyclo [2.2.2]oct-3-yl]2,3,3a,4,5,6-hexahydro-1-oxo-1H-benz[de]isoquinoline hydrochloride. The empirical formula is C19H24N2O.HCl, with a molecular weight of 332.87. Palonosetron hydrochloride exists as a single isomer and has the following structural formula:
 |
Netupitant is white to off-white crystalline powder. It is freely soluble in toluene and acetone, soluble in isopropanol and ethanol, and very slightly soluble in water.
Palonosetron hydrochloride is a white to off-white crystalline powder. It is freely soluble in water, soluble in propylene glycol, and slightly soluble in ethanol and 2-propanol.
Each AKYNZEO capsule is composed of one white-caramel hard gelatin capsule which contains three tablets each containing 100 mg netupitant and one gelatin capsule containing 0.5 mg palonosetron (equivalent to 0.56 mg palonosetron hydrochloride). The inactive ingredients are butylated hydroxyanisole (BHA), croscarmellose sodium, gelatin, glycerin, magnesium stearate, microcrystalline cellulose, mono-and di-glycerides of capryl/capric acid, polyglyceryl dioleate, povidone K-30, purified water, red iron oxide, silicon dioxide, sodium stearyl fumarate, sorbitol, sucrose fatty acid esters, titanium dioxide and yellow iron oxide. It may contain traces of medium-chain triglycerides, lecithin, and denatured ethanol.
AKYNZEO (235 mg fosnetupitant/0.25 mg palonosetron) for injection is a combination product of fosnetupitant, a prodrug of netupitant, which is a substance P/neurokinin 1 (NK-1) receptor antagonist, and palonosetron hydrochloride, a serotonin-3 (5-HT3) receptor antagonist.
Fosnetupitant chloride hydrochloride is chemically described as 2-(3,5-bistrifluoromethylphenyl)-N-methyl-N-[6-(4-methyl-4-O-methylene-phosphatepiperazinium-1-yl)4-o-tolyl-pyridin-3-yl]-isobutyramide chloride hydrochloride. The empirical formula is C31H36F6N4O5P•Cl•HCl, with a molecular weight of 761.53. Fosnetupitant chloride hydrochloride exists as a single isomer and has the following structural formula:
 |
Fosnetupitant chloride hydrochloride is white to off-white to yellowish solid or powder. Its solubility is pH dependent: at acidic pH (pH 2), its solubility is 1.4 mg/mL; at basic pH (pH 10), its solubility is 11.5 mg/mL.
Palonosetron hydrochloride is described above in this section.
AKYNZEO for injection is available for intravenous infusion, and is supplied as a sterile lyophilized powder in a single-dose vial. Each vial contains 235 mg of fosnetupitant (equivalent to 260 mg fosnetupitant chloride hydrochloride) and 0.25 mg of palonosetron (equivalent to 0.28 mg of palonosetron hydrochloride). The inactive ingredients are edetate disodium (6.4 mg), mannitol (760 mg), sodium hydroxide and/or hydrochloric acid (for pH adjustment).
PATENT
WO 2013082102
https://patents.google.com/patent/WO2013082102A1/un



PATENT
US 20150011510
https://patents.google.com/patent/US20150011510A1/en
- [0159]

- Synthesis of methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine
Step 1:
- [0160]
13.0 g (82.5 mMol) 6-Chloro-nicotinic acid in 65 ml THF were cooled to 0° C. and 206.3 ml (206.3 mMol) o-tolylmagnesium chloride solution (1M in THF) were added over 45 minutes. The solution obtained was further stirred 3 hours at 0° C. and overnight at room temperature. It was cooled to −60° C. and 103.8 ml (1.8 Mol) acetic acid were added, followed by 35 ml THF and 44.24 g (165 mMol) manganese(III) acetate dihydrate. After 30 minutes at −60° C. and one hour at room temperature, the reaction mixture was filtered and THF removed under reduced pressure. The residue was partitioned between water and dichloromethane and extracted. The crude product was filtered on silica gel (eluent: ethyl acetate/toluene/formic acid 20:75:5) then partitioned between 200 ml aqueous half-saturated sodium carbonate solution and 100 ml dichloromethane. The organic phase was washed with 50 ml aqueous half-saturated sodium carbonate solution. The combined aqueous phases were acidified with 25 ml aqueous HCI 25% and extracted with dichloromethane. The organic extracts were dried (Na2SO4) and concentrated under reduced pressure to yield 10.4 g (51%) of 6-chloro-4-o-tolyl-nicotinic acid as a yellow foam. MS (ISN): 246 (M−H, 100), 202 (M-CO2H, 85), 166 (36).
Step 2:
- [0161]
To a solution of 8.0 g (32.3 mMol) 6-chloro-4-o-tolyl-nicotinic acid in 48.0 ml THF were added 3.1 ml (42.0 mMol) thionylchloride and 143 .mu.l (1.8 mMol) DMF. After 2 hours at 50° C., the reaction mixture was cooled to room temperature and added to a solution of 72.5 ml aqueous ammonium hydroxide 25% and 96 ml water cooled to 0° C. After 30 minutes at 0° C., THF was removed under reduced pressure and the aqueous layer was extracted with ethyl acetate. Removal of the solvent yielded 7.8 g (98%) 6-chloro-4-o-tolyl-nicotinamide as a beige crystalline foam. MS (ISP): 247 (M+H+, 100).
Step 3:
- [0162]
1.0 g (4.05 mMol) 6-Chloro-4-o-tolyl-nicotinamide in 9.0 ml 1-methyl-piperazine was heated to 100° C. for 2 hours. The excess N-methyl-piperazine was removed under high vacuum and the residue was filtered on silica gel (eluent: dichloromethane) to yield 1.2 g (95%) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-nicotinamide as a light yellow crystalline foam.
- [0163]
MS (ISP): 311 (M+H+, 100), 254 (62).
Step 4:
- [0164]
A solution of 0.2 g (0.6 mMol) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-nicotinamide in 1.0 ml methanol was added to a solution of 103 mg (2.6 mMol) sodium hydroxide in 1.47 ml (3.2 mMol) NaOCl (13%) and heated for 2 hours at 70° C. After removal of methanol, the aqueous layer was extracted with ethyl acetate. The combined organic extracts were dried (Na2SO4), concentrated under reduced pressure and the residue filtered on silica gel (eluent: dichloromethane/methanol 4:1) to yield 100 mg (70%) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-ylamine as a brown resin. MS (ISP): 283 (M+H+, 100), 226 (42).
Step 5:
- [0165]
2.15 mil (11.6 mMol) Sodium methoxide in methanol were added over 30 minutes to a suspension of 0.85 g (4.6 mMol) N-bromosuccinimide in 5.0 ml dichloromethane cooled to −5° C. The reaction mixture was stirred 16 hours at −5° C. Still at this temperature, a solution of 1.0 g (3.1 mMol) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-nicotinamide in 5.0 ml methanol was added over 20 minutes and stirred for 5 hours. 7.1 ml (7.1 mMol) Aqueous HCl 1N and 20 ml dichloromethane were added. The phases were separated and the organic phase was washed with deionized water. The aqueous phases were extracted with dichloromethane, brought to pH=8 with aqueous NaOH 1N and further extracted with dichloromethane. The latter organic extracts were combined, dried (Na2SO4) and concentrated to yield 1.08 g (quant.) [6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-carbamic acid methyl ester as a grey foam.
- [0166]
MS (ISP): 341 (M+H+, 100), 284 (35).
Step 6:
- [0167]
A solution of 0.5 g (1.4 mMol) [6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-carbamic acid methyl ester in 3.0 ml dichloromethane was added over 10 minutes to a solution of 1.98 ml (6.9 mMol) Red-Al®. (70% in toluene) and 2.5 ml toluene (exothermic, cool with a water bath to avoid temperature to go >50° C.). The reaction mixture was stirred 2 hours at 50° C. in CH2Cl2, extracted with ethyl acetate and cooled to 0° C. 4 ml Aqueous NaOH 1N were carefully (exothermic) added over 15 minutes, followed by 20 ml ethyl acetate. The phases were separated and the aqueous phase was extracted with ethyl acetate. The combined organic extracts were washed with deionized water and brine, dried (Na2SO4) and concentrated under reduced pressure to yield 0.37 g (89%) methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine as an orange resin. MS (ISP): 297 (M+H+, 100).
Synthesis of 2-(3,5-bis-Trifluoromethyl-phenyl)-2-methyl-propionyl Chloride
- [0168]

- [0169]
15.0 g (50 mmol) 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionic acid were dissolved in 127.5 ml dichloromethane in the presence of 0.75 ml DMF. 8.76 ml (2 eq.) Oxalyl chloride were added and after 4.5 hours, the solution was rotary evaporated to dryness. 9 ml Toluene were added and the resulting solution was again rotary evaporated, then dried under high vacuum yielding 16.25 g (quant.) of 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride as a yellow oil of 86% purity according to HPLC analysis. NMR (250 MHz, CDCl3): 7.86 (br s, 1H); 7.77, (br s, 2H, 3Harom); 1.77 (s, 6H, 2 CH3).
Synthesis of 2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide (Netupitant)
- [0170]

- [0171]
A solution of 20 g (67.5 mmol) methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine and 17.5 ml (101 mmol) N-ethyldiisopropylamine in 200 ml dichloromethane was cooled in an ice bath and a solution of 24 g (75 mmol)2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride in 50 ml dichloromethane was added dropwise. The reaction mixture was warmed to 35-40° C. for 3 h, cooled to room temperature again and was stirred with 250 ml saturated sodium bicarbonate solution. The organic layer was separated and the aqueous phase was extracted with dichloromethane. The combined organic layers were dried (magnesium sulfate) and evaporated. The residue was purified by flash chromatography to give 31.6 g (81%) of 2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide as white crystals.
- [0172]
M.P. 155-157° C.; MS m/e (%): 579 (M+H+, 100).
Synthesis of 5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-2-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridine 1-oxide
- [0173]

Step 1:
- [0174]
The solution of 6-chloropyridin-3-amine (115 g, 0.898 mol) and (Boc)2O (215.4 g, 0.988 mol) in 900 mL of dioxane was refluxed overnight. The resulting solution was poured into 1500 mL of water. The resulting solid was collected, washed with water and re-crystallized from EtOAc to afford 160 g tert-butyl (6-chloropyridin-3-yl)carbamate as a white solid (Yield: 78.2%).
Step 2:
- [0175]
To the solution of tert-butyl (6-chloropyridin-3-yl)carbamate (160 g, 0.7 mol) in 1 L of anhydrous THF was added n-BuLi (600 mL, 1.5 mol) at −78° C. under N2 atmosphere. After the addition was finished, the solution was stirred at −78° C. for 30 min, and the solution of I2 (177.68 g, 0.7 mol) in 800 mL of anhydrous THF was added. Then the solution was stirred at −78° C. for 4 hrs. TLC indicated the reaction was over. Water was added for quench, and EtOAc was added to extract twice. The combined organic phases were washed with brine, dried over Na2SO4, filtered and purified by flash chromatography to afford 80 g of tert-butyl (6-chloro-4-iodopyridin-3-yl)carbamate as a yellow solid (32.3%).
Step 3:
- [0176]
To the solution of tert-butyl (6-chloro-4-iodopyridin-3-yl)carbamate (61 g, 0.172 mol) in 300 mL of anhydrous THF was added 60% NaH (7.6 g, 0.189 mol) at 0° C. under N2 atmosphere. After the addition was finished, the solution was stirred for 30 min, and then the solution of MeI (26.92 g, 0.189 mol) in 100 mL of dry THF was added. Then the solution was stirred at 0° C. for 3 hrs. TLC indicated the reaction was over. Water was added for quench, and EtOAc was added to extract twice. The combined organic phases were washed with brine, dried over Na2SO4, filtered and concentrated to afford 63 g of crude tert-butyl (6-chloro-4-iodopyridin-3-yl)(methyl)carbamate used into the following de-protection without the further purification.
Step 4:
- [0177]
To the solution of tert-butyl (6-chloro-4-iodopyridin-3-yl)(methyl)carbamate (62.5 g, 0.172 mol) in 500 mL of anhydrous DCM was added 180 mL of TFA. Then the solution was stirred at room temperature for 4 hrs. Concentrated to remove the solvent, and purified by flash chromatography to afford 45.1 g 6-chloro-4-iodo-N-methylpyridin-3-amine as a yellow solid (Yield: 97.3%).
Step 5:
- [0178]
To the solution of 6-chloro-4-iodo-N-methylpyridin-3-amine (40.3 g, 0.15 mol) and 2-methylbenzene boric acid (24.5 g, 0.18 mol) in 600 mL of anhydrous toluene was added 400 mL of 2 N aq. Na2CO3 solution, Pd(OAc)2 (3.36 g, 15 mmol) and PPh3 (7.87 g, 0.03 mmol). The solution was stirred at 100° C. for 2 hrs. Cooled to room temperature, and diluted with water. EtOAc was added to extract twice. The combined organic phases were washed with water and brine consecutively, dried over Na2SO4, concentrated and purified by flash chromatography to afford 19 g 6-chloro-N-methyl-4-(o-tolyl)pyridin-3-amine as a white solid (Yield: 54.6%).
Step 6:
- [0179]
To the solution of 6-chloro-N-methyl-4-(o-tolyl)pyridin-3-amine (18.87 g, 81.3 mmol) and DMAP (29.8 g, 243.9 mmol) in 200 mL of anhydrous toluene was added the solution of 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride (28.5 g, 89.4 mmol) in toluene under N2 atmosphere. The solution was heated at 120° C. for 23 hrs. Cooled to room temperature, poured into 1 L of 5% aq. NaHCO3 solution, and extracted with EtOAc twice. The combined organic phases were washed by water and brine consecutively, dried over Na2SO4, filtered and purified by flash chromatography to afford 35 g 2-(3,5-bis(trifluoromethyl)phenyl)-N-(6-chloro-4-(o-tolyl)pyridin-3-yl)-N,2-dimethylpropanamide as a white solid (Yield: 83.9%).
Step 7:
- [0180]
To the solution of 2-(3,5-bis(trifluoromethyl)phenyl)-N-(6-chloro-4-(o-tolyl)pyridin-3-yl)-N,2-dimethylpropanamide (5.14 g, 10 mmol) in 60 mL of DCM was added m-CPBA (6.92 g, 40 mmol) at 0° C. under N2 atmosphere. Then the solution was stirred overnight at room temperature. 1 N aq. NaOH solution was added to wash twice for removing the excess m-CPBA and a side product. The organic phase was washed by brine, dried over Na2SO4, filtered and concentrated to afford 5.11 g of crude 5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-2-chloro-4-(o-tolyl)pyridine 1-oxide as a white solid (Yield: 96.4%).
Step 8:
- [0181]
To the solution of crude 5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-2-chloro-4-(o-tolyl)pyridine 1-oxide (5.1 g, 9.62 mmol) in 80 mL of n-BuOH was added N-methylpiperazine (7.41 g, 74.1 mmol) under N2 atmosphere. Then the solution was stirred at 80° C. overnight. Concentrated and purified by flash chromatography to afford 4.98 g 5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-2-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridine 1-oxide as a white solid (Yield: 87.2%). 1HNMR (CDCl3, 400 MHz) δ 8.15 (s, 1H), 7.93 (s, 1H), 7.78 (s, 2H), 7.38 (m, 2H), 7.28 (m, 1H), 7.17 (m, 1H), 7.07 (s, 1H), 5.50 (s, 3H), 2.72 (d, J=4.4 Hz, 4H), 2.57 (m, 3H), 2.40 (s, 3H), 2.23 (s, 3H), 1.45˜1.20 (m, 6H).
Synthesis of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-1-oxido-4-(o-tolyl)pyridin-2-yl)-1-methylpiperazine 1-oxide
- [0182]

- [0183]
To a solution of 5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-2-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridine 1-oxide (3 g, 5.05 mmol) and NaHCO3 (0.354 g, 12.66 mmol) in 60 mL of MeOH and 15 mL of H2O were added potassium monopersulfate triple salt (1.62 g, 26.25 mmol) at room temperature during 15 min. After stirring for 4 hrs at room temperature under N2 atmosphere, the reaction mixture was concentrated in vacuo and purified by flash chromatography (eluent: MeOH). The product was dissolved into DCM, the formed solid was filtered off, and the solution was concentrated under reduced pressure to afford 1.77 g 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-1-oxido-4-(o-tolyl)pyridin-2-yl)-1-methylpiperazine 1-oxide as a white solid (Yield: 57.4%). 1HNMR (CDCl3, 400 MHz) δ 8.06 (s, 1H), 7.78 (s, 1H), 7.60 (s, 2H), 7.37˜7.20 (m, 4H), 6.81 (s, 1H), 3.89 (s, 21H), 3.74 (m, 4H), 3.31 (m, 5H), 2.48 (s, 3H), 2.18 (s, 3H), 1.36 (s, 6H).
Synthesis of 1-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-4-methylpiperazine 1,4-dioxide
- [0184]

- [0185]
To the solution of 2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide (11.1 g, 19.2 mmol) in 75 ml of Methanol was added sodium bicarbonate (3.38 g, 40.3 mmol) dissolved in 20 ml of water. Then Oxone (14.75 g, 48.0 mmol) was added to the stirred solution at room temperature in 3-4 portions. The suspension was heated for 4 h at 50° C. After filtration of the salts (washed with 3×8 ml of methanol), the solvent has been evaporated under reduced pressure and substituted by DCM (30 ml). The organic phase was washed with water (5×30 ml), dried over Na2SO4, filtered, concentrated and purified by precipitation in toluene to afford 9.3 g 1-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-4-methylpiperazine 1,4-dioxide as a white solid (Yield: 80%). 1H-NMR (CDCl3, 400 MHz, at 333K) δ 8.27 (s, 2H), 7.75 (s, 1H), 7.63 (s, 2H), 7.26˜7.19 (m, 2H), 7.14 (t, 1H, J=7.4 Hz), 7.09 (d, 1H, J=7.4 Hz), 4.93 (t, 2H, J=11.6 Hz), 4.70 (t, 2H, J=11.6 Hz), 4.12 (d, 2H, J=10.7 Hz), 3.84 (s, 3H), 3.50 (d, 2H, J=10.3 Hz), 2.47 (s, 3H), 2.12 (s, 3H), 1.40 (s, 6H).
Synthesis (A) of di-tert-butyl (chloromethyl)phosphate
- [0186]

- [0187]
Di-tert-butyl phosphohite (40.36 mmole) was combined with potassium bicarbonate (24.22 mmole) in 35 ml of water. The solution was stirred in an ice bath and potassium permanganate (28.25 mmole) was added in three equal portions over one hour’s time. The reaction as then allowed to continue at room temperature for an additional half hour.
- [0188]
Decolorizing carbon (600 mg) was then incorporated as the reaction was heated to 60° C. for 15 minutes. The reaction was then vacuum filtered to remove solid magnesium dioxide. The solid was washed several times with water. The filtrate was then combined with one gram of decolorizing carbon and heated at 60° C. for an additional twenty minutes. The solution was again filtered to yield a colorless solution, which was then evaporated under vacuum to afford crude Di-tert-butyl phosphate potassium salt. Di-tert-butyl phosphate potassium salt (5 g, 20.14 mmole) was dissolved in methanol (15 g): to this solution at 0° C. a slight excess of concentrated HCl is slowly added with efficient stirring at 0° C. The addition of acid causes the precipitation of potassium chloride. The solid is then filtered and washed with methanol. The compound in the mother liquor is then converted to the ammonium form by adding an equal molar amount of tetramethylammonium hydroxide (3.65 g, 20.14 mmole) while keeping the reaction cooled by a salt/ice bath with efficient stirring. The resulting clear solution is placed under reduced pressure to give the crude product. To the tetramethylammonium di-tert-butyl-phosphate dissolved in refluxing dimethoxyethane is then added 4.3 grams of chloroiodomethane (24.16 mmole) and stirred for 1-2 hours. The reaction is then filtered and the filtrate is placed under reduced pressure to concentrate the solution in DME. The chloromethyl di-tert-butyl phosphate 12-16% in DME is used in the synthesis of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-((phosphonooxy)methyl)piperazin-1-ium without further purifications (60% yield): 1HNMR (CD3OD, 300 MHz) δ 1.51 (s, 12H), 5.63 (d, 2H, J=14.8). 31P-NMR (CD3OD, 300 MHz) δ −11.3 (s, 1P).
Synthesis (B) of di-tert-butyl (chloromethyl)phosphate
- [0189]

- [0190]
Di-tert-butyl phosphate potassium salt (5 g, 20.14 mmole) is dissolved in methanol (15 g): to this solution at 0° C. a slight excess of concentrated HCl is slowly added with efficient stirring at 0° C. The addition of acid causes the precipitation of potassium chloride. The solid is then filtered and washed with methanol. The compound in the mother liquor is then converted to the ammonium form by adding an equal molar amount of tetrabuthylammonium hydroxide 1 M in methanol (20.14 mmole) while keeping the reaction cooled at 0° C. with efficient stirring. The resulting clear solution is placed under reduced pressure to give the intermediate product. The tetrabuthylammonium di-tert-butyl-phosphate dissolved in acetone is then added dropwise to 53.3 grams of chloroiodomethane (302.1 mmole) and stirred at 40° C. for 1-2 hours. The solvent and excess of chloroiodomethane are distilled off, the reaction mass suspended in TBME and then filtered. The filtrate is washed by a saturated solution of sodium bicarbonate and water and then placed under reduced pressure to substitute the solvent by acetone, i.e., to remove the solvent after which it is replaced with acetone. The chloromethyl di-tert-butyl phosphate 7-20% in acetone is used in the next step without further purifications (70-80% yield): 1H-NMR (CD3OD, 300 MHz) δ 1.51 (s, 12H), 5.63 (d, 2H, J=14.8). 31P-NMR (CD3OD, 300 MHz) δ −11.3 (s, 1P).
Stability studies of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-((phosphonooxy)methyl)piperazin-1-ium salts
- [0191]
In order to further improve the stability and solubility of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-((phosphonooxy)methyl)piperazin-1-ium, a variety of its derivative salts were synthesized and tested. Their synthesis employed either a) neutralization of the dried diacid phosphate species and its corresponding base salts or b) a direct acid deprotection starting from the dried di(tert-butyl)-protected phosphate species. Neutralization was performed with L-histidine, magnesium salt, N-methyl-D-glucamine (dimeglumine), and L-lysine. Both procedures were tried in the synthesis of citric derivatives whereas with other acids the direct deprotection reaction was used. The figures below show the most relevant structures.
-

- [0192]
When the parent acid species was not stored in dry condition it was found to undergo over 8% degradation in the first week and over 65% degradation in the first six months. When the dried parent acid species was held at 30° C. in air it underwent 0.05% degradation in the first 7 days and at total of 7.03% degradation in six months. When the dried parent acid species was held under argon at room temperature it underwent up to 0.13% degradation in the first 7 days but then was essentially stable for six months. Results for various derivative salts are shown in Table 1 below.
-
TABLE 1 Representative Degradation Results for Salts Purity A % Solvents Additives Yield % HPLC Comments MeOH L-Histidine, 2 eq. 26.6% 95.94% Degradation: +0.70% in 6 days (in air) +0.46% in 6 days (in argon) MeOH Mg(OH)2, 2 eq. 48.6% 94.11% Degradation: +0.81% in 6 days (in air) +0.29% in 6 days (in argon) MeOH + Citric acid, 2 eq. N.A. 94.40% From protected species. DCM, 1:1 MeOH 1. HCl dioxane, 4 eq. >90% 94.50% From protected species. 2. Ca(OH)2 MeOH H3PO4, 85%, 2 eq. >90% 98.81% From protected species and retains 0.39% of that species. MeOH HBr, 48%, 4 eq. 84.6% 96.11% From protected species. Product degrades rapidly, MeOH + CH3SO3H N.A. 61.54% From protected species. DCM, Product NOT stable: contains 1:4 32.45% decomposition species. MeOH NaH2PO4, 4 eq. N.A. n.d. Only 1.27 of parent species formed. Poor reaction. MeOH N-methyl-D- N.A. 96.88% Degradation: glucamine +0.87% in 6 days (in air) (Meglumine), 2 eq. +1.52% in 11 days (in argon) MeOH N-methyl-D- >99% 97.42% Degradation: glucamine +0.77% in 6 days (in air) (Meglumine), 1 eq. +0.83% in 7 days (in argon) MeOH+ 1. NaOH, 3 eq 96.5% 97.49% Degradation: DCM, 2. Citric acid, 1 eq. +0.09% in 2 days (in argon) 5:2 +0.59% in 89 days (in argon) MeOH+ 1. NaOH, 3 eq. 93.8% 97.46% Degradation: DCM, 2. Fumaric acid, 1 eq. +1.95% in 14 days (in air) 5:2 +1.80% in 12 days (in argon) MeOH L-lysine, 1 eq. >99% 97.62% Degradation: +0.69% in 14 days (in air) +0.48% in 12 days (in argon)
- [0194]
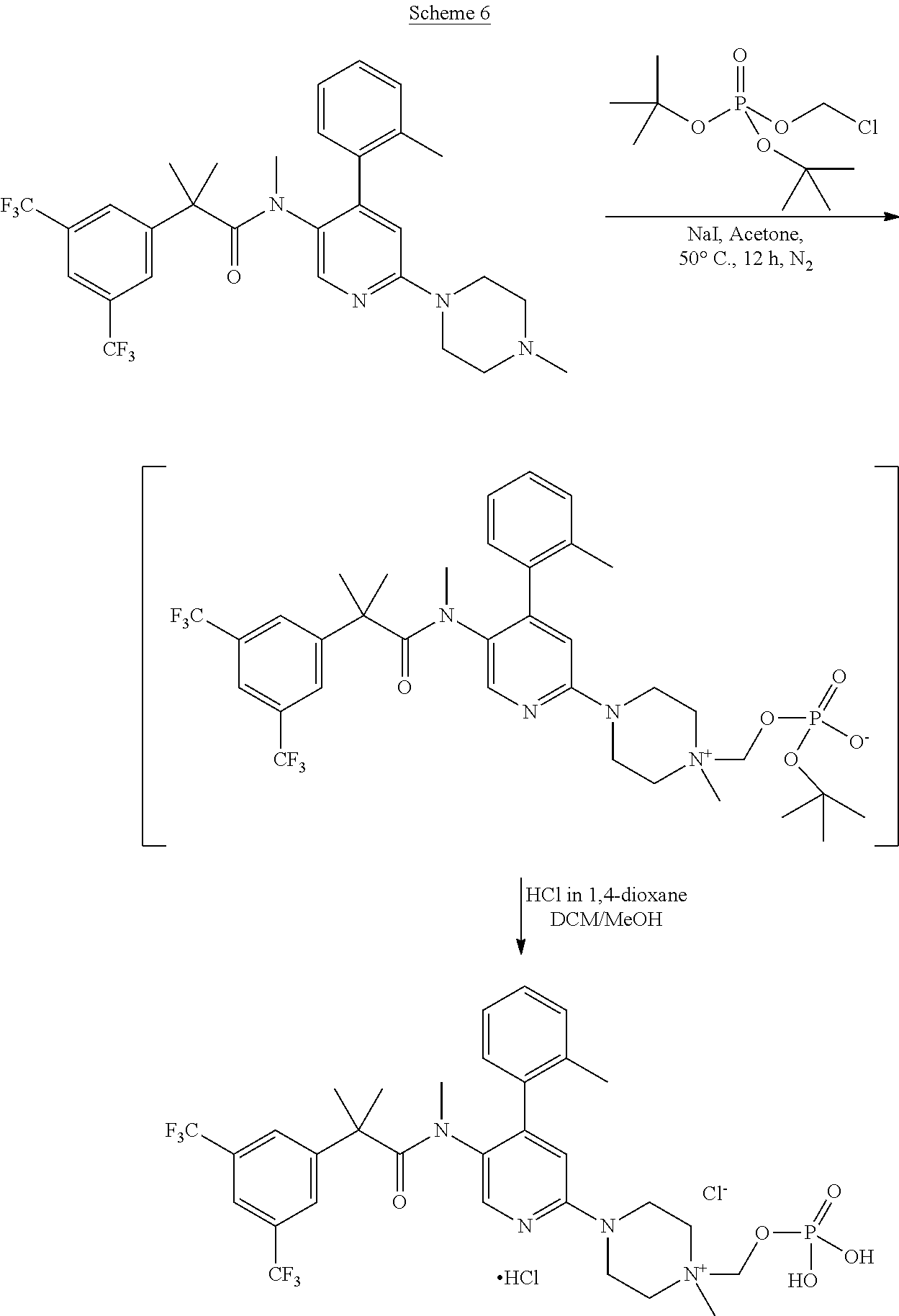
- [0195]
The solution of chloromethyl di-tert-butyl phosphate in DME (250 g from a 10% solution, 96.64 mmole) was evaporated under reduced pressure until the formation of pale yellow oil, dissolved then at 50° C. with 318 ml of Acetonitrile. 17.2 g (80.54 mmole) of 1,8-bis(dimethylamino)naphtalene and 46.6 g (80.54 mmole) of 2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide were added and the solution heated at 90° C. for at least 12 h. After the addition of 75 g of isopropylether, the precipitated crude product was cooled at room temperature, filtered and washed with acetonitrile, isopropylether/acetone, 3:1 and isopropylether, and dried under reduced pressure to afford 20-33 g of the 4-(5-{2-[3,5-bis(trifluoromethyl)phenyl]-N,2-dimethylpropanamido}-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-{[(tert-butoxy)phosphoryl]oxymethyl}piperazin-1-ium as white solid (Yield: 30-50%). 1H-NMR (CD3OD, 400 MHz) δ 7.98 (s, 1H), 7.86 (s, 1H), 7.76 (s, 2H), 7.33-7.10 (m, 4H), 6.80 (s, 1H), 5.03 (d, 2H, JPH=8.5 Hz), 4.52 (s, 2H), 4.13 (m, 2H), 3.83 (m, 2H), 3.69 (m, 2H), 3.52 (m. 2H), 3.23 (s, 3H), 2.53 (s, 3H), 2.18 (s, 3H), 1.46 (s, 18H), 1.39 (s, 6H). 31P-NMR (CD3OD, 161 MHz) δ −5.01 (s, 1P). To 20 g (23.89 mmole) of the 4-(5-{2-[3,5-bis(trifluoromethyl)phenyl]-N,2-dimethylpropanamido}-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-{[(tert-butoxy)phosphoryl]oxymethyl}piperazin-1-ium dissolved in 180 g of methanol and 400 g of dichloromethane was added HCl 4M in dioxane (18.8 g, 71.66 mmole) and the solution was heated for 3 h at reflux. After the addition of 200 g of dioxane, DCM and methanol were distilled under reduced pressure until precipitation of the product, which was filtered and washed with isopropylether (100 g), acetone (30 g) and pentane (2×60 g). The product was finally dried under reduced pressure at 55° C. to afford 15-17 g of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-((phosphonooxy)methyl)piperazin-1-ium chloride hydrochloride as white solid (Yield: 88-93%). 1H-NMR (CD3OD, 400 MHz) δ 7.02 (s, 1H), 7.87 (s, 1H), 7.74 (s, 2H), 7.33-7.40 (m, 2H), 7.27 (m, 1H), 7.21 (s, 1H), 7.16 (d, 1H, J=8.2 Hz), 5.27 (d, 2H, JPH=7.9 Hz), 4.29 (m, 2H), 4.05 (m, 2H), 3.85 (m, 2H), 3.74 (m, 2H), 3.35 (s, 3H), 2.62 (s, 3H), 2.23 (s, 3H), 1.38 (s, 6H). 31P-NMR (CD3OD, 161 MHz) δ −2.81 (t, 1P, JPH=7.9 Hz).
- Synthesis (A) of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-((phosphonooxy)methyl)piperazin-1-ium chloride hydrochloride
Synthesis (B) of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-((phosphonooxy)methyl)piperazin-1-ium chloride hydrochloride
- [0196]

- [0197]
To the solution of chloromethyl di-tert-butyl phosphate in Acetone (22.1 g from a 10% solution, 85.58 mmole), 15.5 g (103.24 mmole) of sodium iodide and 33.0 g (57.00 mmole) of netupitant were added and the solution heated at 50° C. for at 6-16 h. The precipitated salts were filtered off, the acetone distilled under reduced pressure and the crude product dissolved in 43.0 g of methanol and 43.0 g 1,4-dioxane. 12.6 g of HCl 4M in dioxane (113.85 mmole) were added, and then methanol is distilled off at 40° C. under reduced pressure. The solution is cooled at 5° C. and stirred at 5° C. for at least 2 h at 5° C. The product was isolated by filtration, purified by additional slurry in acetone (238 g), and filtered and washed with acetone (47 g) and pentane (2×72 g).
- [0198]
The product was finally dried under reduced pressure at 60° C. to afford 22-30 g of white-yellowish solid (Yield: 50-70%)
- [0199]
1H-NMR (CD3OD, 400 MHz) δ 7.02 (s, 1H), 7.87 (s, 1H), 7.74 (s, 2H), 7.33-7.40 (m, 2H), 7.27 (m, 1H), 7.21 (s, 1H), 7.16 (d, 1H, J=8.2 Hz), 5.27 (d, 2H, JPH=7.9 Hz), 4.29 (m, 2H), 4.05 (m, 2H), 3.85 (m, 2H), 3.74 (m, 2H), 3.35 (s, 3H), 2.62 (s, 3H), 2.23 (s, 3H), 1.38 (s, 6H). 31P-NMR (CD3OD, 161 MHz) δ −2.81 (t, IP, JPH=7.9 Hz).
PATENT
US 8,426,450
PATENT
US 9,403,772
SYN
https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/chem.201901840
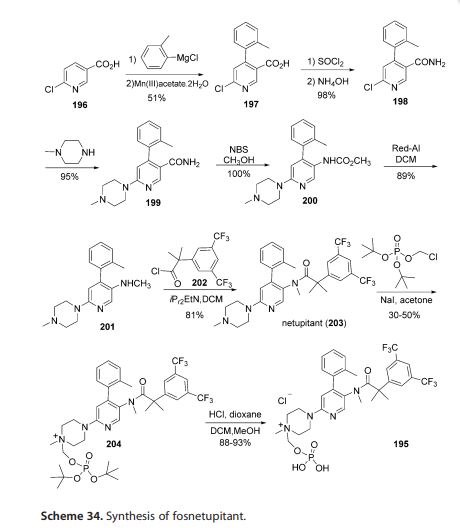
The synthesis of fosnetupitant (195) was developed by the Swiss company Helsinn (Scheme 34).[58] The synthesis started with the reaction of 6-chloronicotinic acid (196) with o-tolylmagnesium chloride followed by manganese(III) acetate to give acid derivative 197. This was converted to amide 198 after reaction with thionyl chloride and ammonium hydroxide. Next, reaction with N-methylpiperazine furnished intermediate 199, which was then transformed into carbamate 200 after reaction with NBS in methanol. Reduction with Red-Al followed by acylation with acyl chloride 202 afforded netupitant (203).
Finally, reaction with di-tert-butyl chloromethyl phosphate followed by the removal of the tert-butyl groups by treatment with HCl in dioxane afforded fosnetupitant (195).
L. Fadini, P. Manini, C. Pietra, C. Giuliano, E. Lovati, R. Cannella, S. Venturini, V. J. Stella, WO 082102 A1, 2013.

SYN
Fosnetupitant chloride HCl

Fosnetupitant is a neurokynin-1 (“NK-1”) antagonist under development by Helsinn Healthcare SA, Lugano/Pazzallo Switzerland, for the treatment of chemotherapy induced nausea and vomiting. The compound is known chemically as 4-(5-(2-(3,5- bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-l-methyl- 1 -((phosphonooxy)methyl)piperazin- 1 -ium, and has the following chemical structure in its acidic/free base form:

[004] The chloride monohydrochloride salt, and a method for its preparation, is described in WO 2013/082102. The chemical structure for this salt is reported as follows:

[005] The molecule can be challenging to manufacture, particularly in a highly pure crystalline form in a commercially acceptable yield. Solvents used in the manufacture of the product pose special challenges. Prior art processes have removed these solvents via evaporative techniques, which can degrade the fosnetupitant due to the excessive heat.
EXAMPLES
[089] In all the examples reported, unless otherwise reported, the starting compound was Form I of the chloride hydrochloride salt of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)- N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-l-methyl-l – ((phosphonooxy)methyl)piperazin-l-ium, produced substantially according to the methods described in WO 2013/082102.
EXAMPLE 1 : CHARACTERIZATION OF FOSNETUPITANT
1 . Experimental Methods
1.1 Solubility
[090] The solubility of the starting compound was determined in 25 pharmaceutically acceptable solvents (class II and III) of differing polarity. The procedure was as follows:
[091] Approximately 20 mg of material was weighed out into each glass vial.
[092] 5 volume aliquots of each solvent were added separately with stirring (i.e. 1 volume = 20 μΐ; hence, 5 volume = 100 μΐ (5 x 20 μΐ)).
[093] The mixture was stirred at RT for 5- 10 minutes. Visual checks were then made for solubility.
[094] If no solubility was achieved then steps (ii) and (iii) were repeated until either the solubility was achieved or the 50 volume aliquots of that solvent were added.
[095] Solubility was then approximated.
[096] Solubility was finally checked at the elevated temperature (40°C).
1.2 Polymorph Screen (including slurry studies)
[097] Using the information from the solubility study, the compound was slurried in the solvents outlined in Table I and two more mixtures of water/ MeOH (10:90) and water/ Acetone (1 :20) respectively with temperature cycling between 40°C and RT (4 hour periods at each temperature) over 48 hours. After the slurries the resulting solids were isolated and analyzed by Raman and XRPD (where enough material was available) for any change in physical form.
[098] The compound was also dissolved in the listed solvents and two more mixtures of water/organic solvent to yield saturated solutions, and crystallization was induced by: crash cooling (at ca. -1 8°C); evaporation (at RT); and addition of an anti-solvent. Solid materials generated were then isolated and examined by Raman and XRPD (where enough material was available).
1.3 Scale-up of any new polymorphic forms
[099] Any new potential polymorphic forms of the Form I fosnetupitant were then scaled-up to ~500mg level for further characterizations by PLM, SEM, DSC, TGA, GVS (XRPD post GVS) and NMR. Further studies of conversion between each polymorphic form were also performed. From this information, an understanding of the polymorphic space was achieved.
Synthetic Reference
Fadini, Luca; Manini, Peter; Pietra, Claudio; Giuliano, Claudio; Lovati, Emanuela; Cannella, Roberta; Venturini, Alessio; Stella, Valentino. (Assignee: Helsinn Healthcare SA, Switz). Substituted 4 – phenyl – pyridines for the treatment of nk-1 receptor related diseases. WO2013082102 (2013).
//////////Fosnetupitant, 07-PNET, Фоснетупитант , فوسنيتوبيتانت , 磷奈匹坦 , FDA 2014, EMA 2015