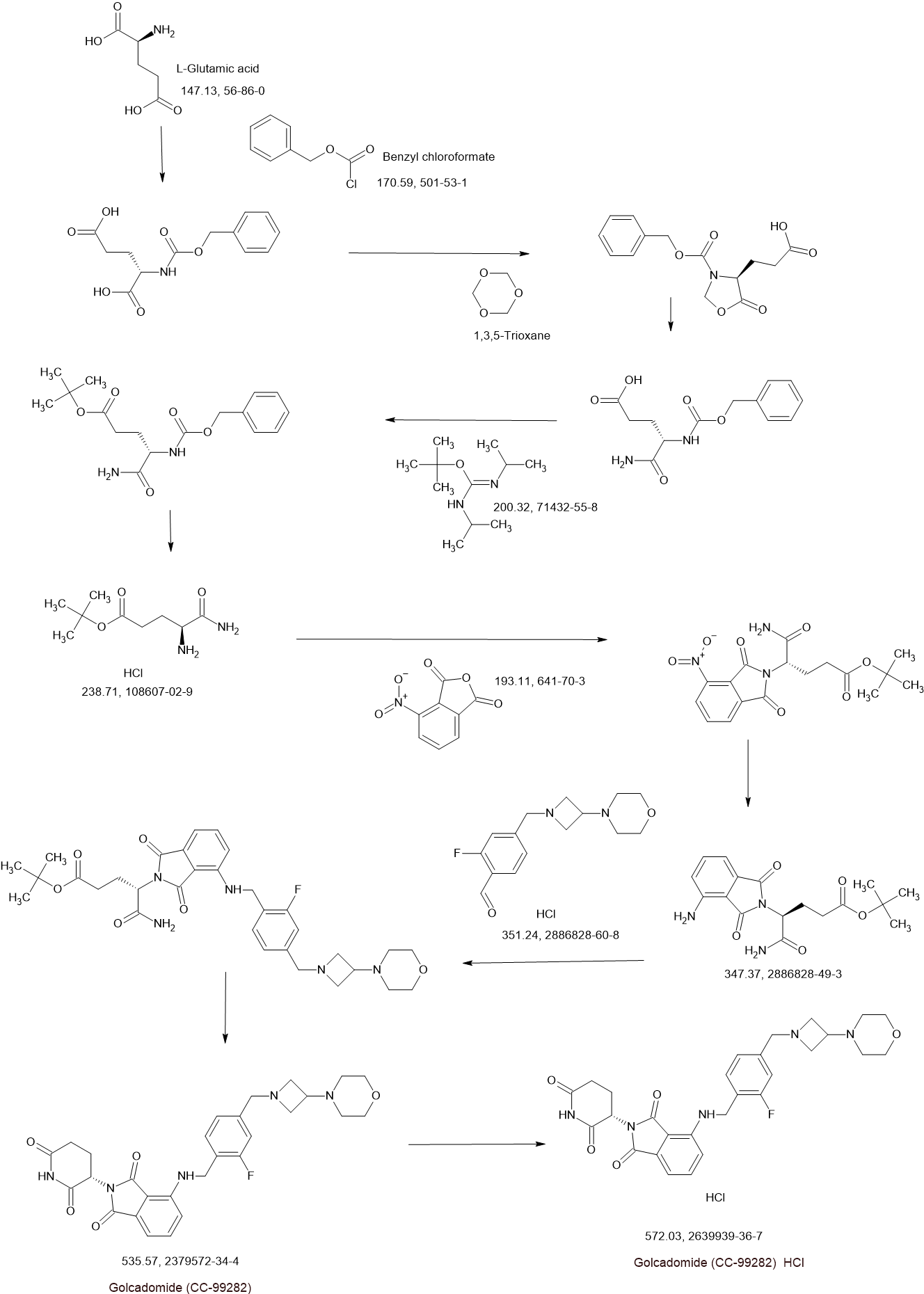
Golcadomide (CC-99282)
| Molecular Weight | 535.57 |
| Formula | C28H30FN5O5 |
| CAS No. | 2379572-34-4 |
(S)-2-(2,6-Dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline-1,3-dione
2-[(3S)-2,6-dioxopiperidin-3-yl]-4-[[2-fluoro-4-[(3-morpholin-4-ylazetidin-1-yl)methyl]phenyl]methylamino]isoindole-1,3-dione
- 2-[(3S)-2,6-Dioxo-3-piperidinyl]-4-[[[2-fluoro-4-[[3-(4-morpholinyl)-1-azetidinyl]methyl]phenyl]methyl]amino]-1H-isoindole-1,3(2H)-dione (ACI)
- (S)-2-(2,6-Dioxopiperidin-3-yl)-4-[[2-fluoro-4-[(3-morpholinoazetidin-1-yl)methyl]benzyl]amino]isoindoline-1,3-dione
- CC 1007548
- CC 99282
- CC-99282
- CC1007548
- Golcadomide
- WHO 12305
Golcadomide HCl, 2639939-36-7
Chemical Name: CC-99282 Hydrochloride; 2-[(3S)-2,6-Dioxopiperidin-3-yl]-4-[[2-fluoro-4-[(3-morpholin-4-ylazetidin-1-yl)methyl]phenyl]methylamino]isoindole-1,3-dione hydrochloride
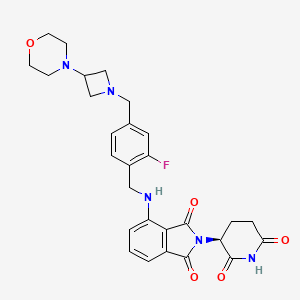
Novel potent and orally active cereblon (CRBN) E3 ligase modulator
Ref: https://www.sciencedirect.com/science/article/abs/pii/S0268960X22000686
Golcadomide is a modulator of the E3 ubiquitin ligase complex containing cereblon (CRL4-CRBN E3 ubiquitin ligase), with potential immunomodulating and antineoplastic activities. Upon administration, golcadomide specifically binds to cereblon (CRBN), thereby affecting the ubiquitin E3 ligase activity, and targeting certain substrate proteins for ubiquitination. This induces proteasome-mediated degradation of certain transcription factors, some of which are transcriptional repressors in T-cells. This leads to modulation of the immune system, including activation of T-lymphocytes, and downregulation of the activity of other proteins, some of which play key roles in the proliferation of certain cancer cell types. CRBN, the substrate recognition component of the CRL4-CRBN E3 ubiquitin ligase complex, plays a key role in the ubiquitination of certain proteins.
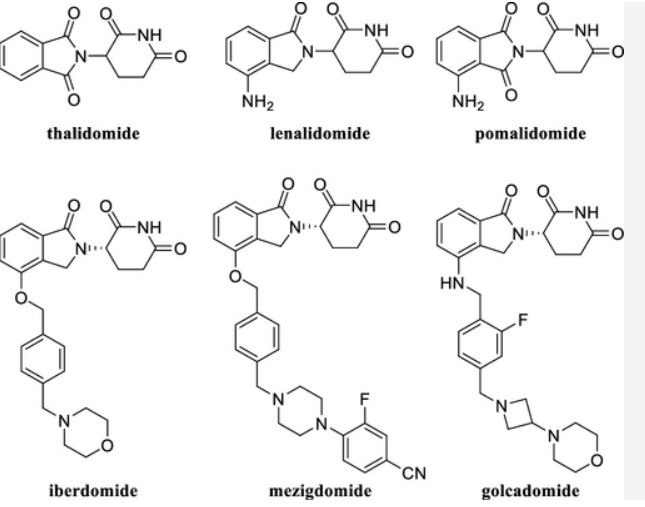
SCHEME
SEE END OF PAGE
PATENT
US20220324855
https://patentscope.wipo.int/search/en/detail.jsf?docId=US376433363&_cid=P22-LR3AIE-86638-1
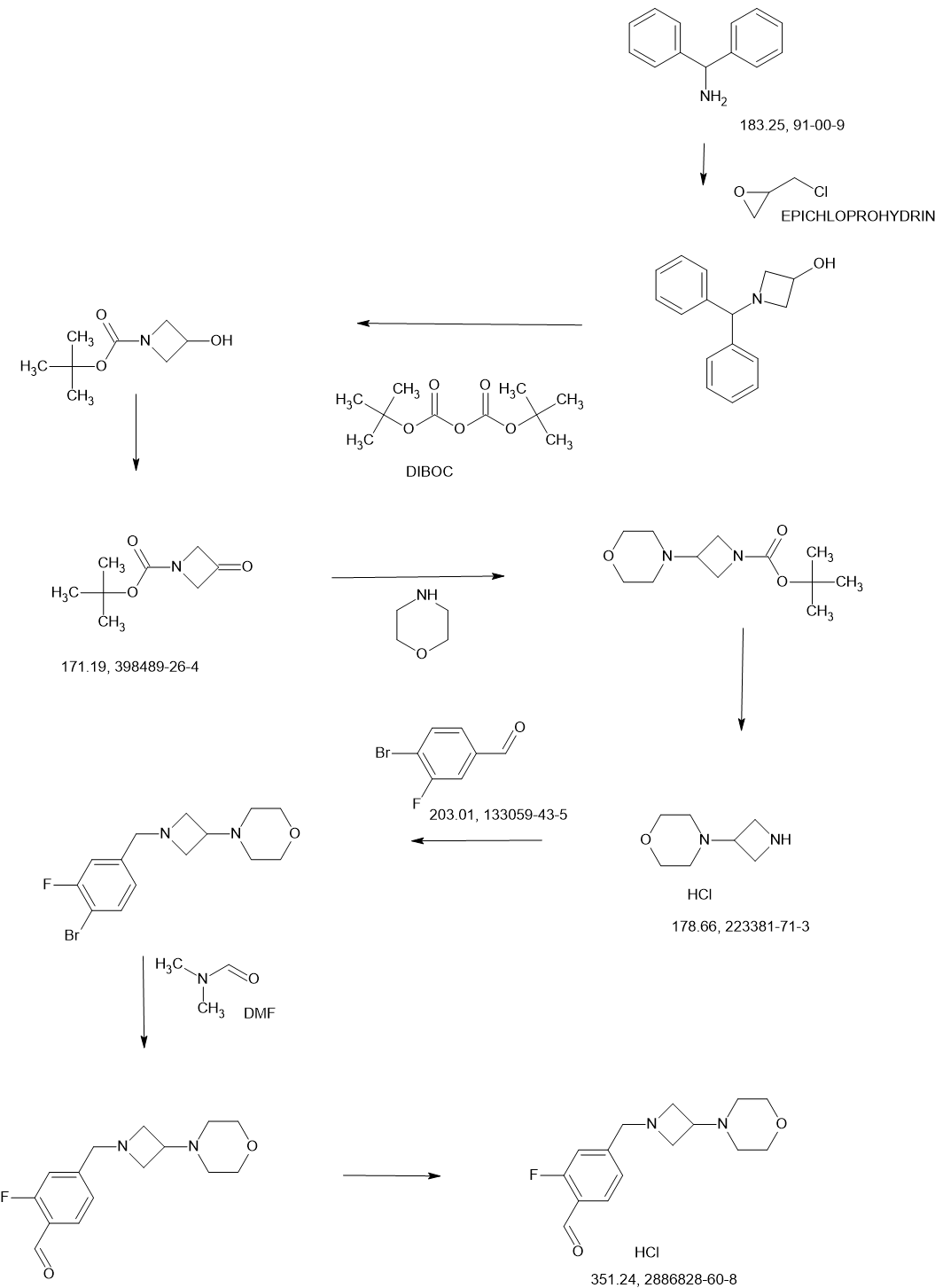
Example 1: Synthesis of (S)-2-(2,6-Dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline-1,3-dione (Compound 1)
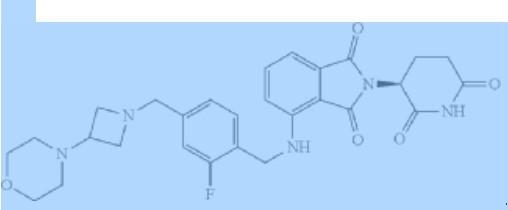
PATENT
WO2022271557
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022271557&_cid=P22-LR3A02-83684-1
Example 1: Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3- morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione
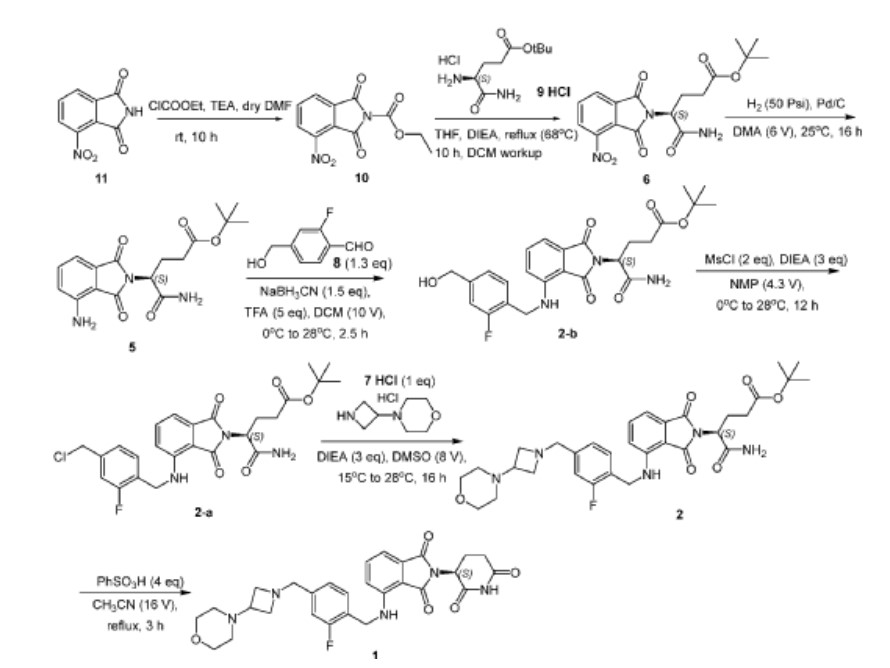
[00242] Synthesis of Ethyl 4-nitro-1,3-dioxo-isoindoline-2-carboxylate (Compound 10): To a solution of 4-nitroisoindoline-1,3-dione (Compound 11, 440 g, 2.29 mol) and TEA (262 g, 2.59 mol, 359 mL) in dry DMF (2.2 L) was cooled to 0 °C and ethyl chloroformate (313 g, 2.89 mol, 275 mL) was added dropwise over 5 minutes. The reaction mixture was stirred at 22 °C for 10 hours. The mixture was slowly added to chilled water (10 L) and the resulting suspension stirred for 5 minutes. The suspension was filtered and the filter cake was washed with water (1 L). The solid was dissolved with ethyl acetate (5 L) and the organic phase was washed with aqueous HCl (1 M, 1 L), water (2 L) and brine (2 L). The organic phase was dried over sodium sulfate , filtered and concentrated to give Compound 10 (360 g, 59%) as a white solid. 1 H NMR (400 MHz CDCl 3 ) δ ppm 8.24 (d, J = 7.6 Hz, 1H), 8.19 (d, J = 8.4 Hz, 1H), 8.06-8.02 (m, 1H), 4.49 (q, J = 7.2 Hz, 2H), 1.44 (t, J = 6.8 Hz, 3H).
[00243] Synthesis of tert-Butyl (4S)-5-amino-4-(4-nitro-1,3-dioxo-isoindolin-2-yl)-5-oxo-pentanoate (Compound 6): To a solution of Compound 10 (165 g, 625 mmol) and DIEA (113 g, 874 mmol, 153 mL) in dry THF (1700 mL) was added tert-butyl (4S)-4,5-diamino-5-oxo-pentanoate hydrochloride ( 149 g, 625 mmol) and heated at reflux for 10 hours. The reaction mixture was concentrated under reduced pressure. The resulting residue was diluted with methyl tert-butyl ether (5 L) and stirred at 20 °C for 1 hour. The suspension was filtered and the filter cake was dissolved with DCM (4 L). The organic phase was washed with water (1.5 L x 3), brine (1.5 L) and dried over sodium sulfate . The organic phase was filtered and concentrated under reduced pressure to give a light yellow oil. The oil was diluted with hexane / ethyl acetate (10/1, 2 L) and stirred until a light yellow suspension formed. The suspension was filtered and the filter cake was triturated and concentrated in vacuum to give Compound 6 (175 g, 74%) as a light yellow solid. 1 H NMR (400 MHz CDCl3) δ ppm 8.12 (d, J = 8.0 Hz, 2H), 7.94 (t, J = 8.0 Hz, 1H), 6.48 (s, 1H), 5.99 (s, 1H), 4.84- 4.80 (m, 1H), 2.49-2.44 (m, 2H), 2.32-2.27 (m, 2H), 1.38 (s, 9H).
[00244] Synthesis of tert-Butyl (S)-5-amino-4-(4-amino-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 5): To a suspension of Compound 6 (170.0 g, 450.5 mmol, 1.00 eq) in DMA (1.00 L) was added palladium on carbon (50.0 g, 10% purity) under nitrogen. The suspension was degassed under vacuum and purged with hydrogen gas several times. The mixture was stirred under hydrogen gas (50 psi) at 25 °C for 16 hours. The mixture was filtered and the filtrate was poured into cooled water (3.0 L). The mixture was stirred at 10 °C for 1 hour and filtered. The filter cake was washed with water (700 mL) and dissolved in DCM (1.00 L). The organic phase was dried over sodium sulfate , filtered and concentrated under reduced pressure to give Compound 5 (107 g, 68%) as a green solid. 1 H NMR (400 MHz DMSO-d 6 ) δ ppm 7.52 (s, 1H), 7.43 (dd, J = 8.4, 7.2 Hz, 1H), 7.13 (s, 1H), 6.95-6.99 (m, 2H), 6.42 (s, 2H), 5.75 (s, 1H), 4.47-4.51 (m, 1H), 2.32-2.33 (m, 1H), 2.14-2.20 (m, 3H), 1.32 (s, 9H); HPLC purity, 100.0%; SFC purity, 100.0% ee.
[00245] Synthesis of 2-fluoro-4-(hydroxymethyl)benzaldehyde (Compound 8): To a solution of 4-(((tert-butyldimethylsilyl)oxy)methyl)-2-fluorobenzaldehyde (370.0 g, 1.38 mol, 1.00 eq ) in THF (1.85 L) was added a solution of p-toluenesulfonic acid monohydrate (78.7 g, 413.6 mmol, 0.30 eq) in water (1.85 L) drop-wise at 10 °C. The mixture was stirred at 27 °C for 16 hours. TEA (80 mL) was added drop-wise and stirred for 10 minutes. The organic phase was separated and the aqueous phase was extracted with ethyl acetate (600 mL × 4). The combined
organic phase was washed with brine (1.50 L), dried over anhydrous sodium sulfate , filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give Compound 8 (137.5 g, 76%) as a yellow oil. 1 H NMR (400 MHz CDCl 3 ) δ ppm 10.34 (s, 1H), 7.86 (dd, J = 8.0, 7.2 Hz, 1H), 7.25 (s, 1H), 7.22 (d, J = 4.4 Hz, 1H) , 4.79 (d, J = 6.0 Hz, 2H), 1.91 (t, J = 6.0 Hz, 1H).
[00246] Synthesis of tert-Butyl (S)-5-amino-4-(4-((2-fluoro-4-(hydroxymethyl)benzyl)amino)-1,3-dioxoisoindolin-2-yl)-5- oxopentanoate (Compound 2-b): To a solution of Compound 5 (100.0 g, 287.9 mmol, 1.00 eq) and Compound 8 (57.7 g, 374.3 mmol, 1.30 eq) in dry DCM (1.00 L) was added TFA (164.1 g , 1.44 mol, 5.00 eq) at 0 °C. The reaction mixture was stirred at 28 °C for 2 hours. To the solution was added sodium cyanoborohydride (27.1 g, 431.8 mmol, 1.50 eq) at 0 °C. The mixture was stirred at 28 °C for 30 minutes. The reaction mixture was quenched by addition of MeOH (600 mL) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give Compound 2-b (110.0 g, 74.0%) as a yellow solid. 1 H NMR (400 MHz, DMSO-d6) δ ppm 7.56 (s, 1H), 7.50 (dd, J = 8.4, 7.2 Hz, 1H), 7.34 (t, J = 8.0 Hz, 1H), 7.02-7.18 ( m, 4H), 6.94-7.01 (m, 2H), 4.57 (d, J = 6.0 Hz, 2H), 4.47-4.53 (m, 3H), 2.31-2.35 (m, 1H), 2.15-2.22 (m, 3H), 1.31 (s, 9H); HPLC purity, 94.0%; SFC purity, 100.0% ee.
[00247] Synthesis of tert-butyl (S)-5-amino-4-(4-((4-(chloromethyl)-2-fluorobenzyl)amino)-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 2-a): To a solution of Compound 2-b (100.0 g, 206.0 mmol, 1.00 eq) in NMP (430.0 mL) was added DIEA (79.9 g, 617.9 mmol, 3.00 eq) and MsCl (47.2 g, 411.9 mmol, 2.00 eq) at 0 °C. The ice bath was removed, and the reaction was stirred at 28°C for 10 hours. The reaction was poured into cooled water (<10°C, 2.0 L) and stirred for 10 minutes. The mixture was extracted with methyl tert-butyl ether (750 mL x 3). The combined organic layer was washed with brine (1.25 L), dried over sodium sulfate , filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give Compound 2-a (86.0 g, 81.2%) as a yellow solid.
1 H NMR (400 MHz DMSO-d 6 ) δ ppm 7.55 (s, 1H), 7.50 (dd, J = 8.4, 7.2 Hz, 1H), 7.38 (t, J = 8.0Hz, 1H), 7.31 (dd, J = 10.8, 1.6 Hz, 1H), 7.23 (dd, J = 8.0, 1.6 Hz, 1H), 7.16 (s, 1H), 7.11 (t, J = 6.4 Hz, 1H), 7.00 (d, J = 7.2 Hz, 1H), 6.95 (d, J = 8.4 Hz, 1H), 4.74 (s, 2H), 4.61 (d, J = 6.4 Hz, 2H), 4.49-4.53 (m, 1H), 2.29-2.38 (m , 1H), 2.16-2.25 (m, 3H), 1.30 (s, 9H); HPLC purity, 98.0%; SFC purity, 100.0% ee.
[00248] Synthesis of tert-butyl (S)-5-amino-4-(4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)-1,3- dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 2): To a solution of 4-(azetidin-3-yl)morpholine hydrochloride (Compound 7 HCl, 30.5 g, 170.7 mmol, 1.00 eq) and DIEA (66.2 g, 512.0 mmol, 3.00 eq) in DMSO (350.0 mL) was added to a solution of Compound 2-a (86 g, 170.65 mmol, 1.00 eq) in DMSO (350.0 mL) drop-wise at 15 °C. The reaction mixture was stirred at 28 °C for 16 hours. The reaction mixture was poured into cold half saturated brine (<10°C, 2.5 L) and extracted with ethyl acetate (1.50 L, 1.00 L, 800.0 mL). The combined organic phase was washed with saturated brine (1.50 L), dried over sodium sulfate , filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give Compound 2 (68.3 g, 65.7%) as a yellow solid. 1 H NMR (400 MHz DMSO-d 6 ) δ ppm 7.55 (s, 1H), 7.50 (dd, J = 8.4, 7.2 Hz, 1H), 7.31 (t, J = 8.0 Hz, 1H), 7.16 (s, 1H), 6.94-7.10 (m, 5H), 4.56 (d, J = 6.4 Hz, 2H), 4.49-4.52 (m, 1H), 3.54-3.55 (m, 6H) 3.31-3.32 (m, 3H), 2.81-2.88 (m, 3H), 2.29-2.38 (m, 1H), 2.15-2.25 (m, 7H), 1.30 (s, 9H); HPLC purity, 100.0%; SFC purity, 100.0% ee.
[00249] Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione (Compound 1): A solution of Compound 2 (30.0 g, 49.2 mmol, 1.00 eq) and benzenesulfonic acid (31.1 g, 196.8 mmol, 4.00 eq) in acetonitrile (480.0 mL) was stirred at reflux for 3 hours. The reaction was cooled to 20 °C, poured into cold brine:saturated sodium bicarbonate solution (1:1, <10 °C, 2.0 L) and extracted with ethyl acetate (1.0 L). The organic phase was washed with cold brine:saturated sodium bicarbonate solution (1:1, <10°C, 1.00 L) once more. The combined aqueous phase was extracted with ethyl acetate (500.0 mL x 2). The combined organic phase was washed with cold brine (<10°C, 1.0 L), dried over sodium sulfate , filtered and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography to give Compound 1 (17.5 g, 66.0%) as a yellow solid. 1 H NMR (400 MHz, DMSO-d 6 ) δ ppm 11.10 (s, 1H), 7.54 (t, J = 8.0 Hz, 1H), 7.30 (t, J = 8.0 Hz, 1H), 7.04-7.10 (m , 4H), 7.00 (d, J = 8.4 Hz, 1H), 5.07 (dd, J = 12.8, 5.2 Hz, 1H), 4.58 (d, J = 6.4 Hz, 2H), 3.53-3.55 (m, 6H) , 3.30-3.32 (m, 2H), 2.81-2.89 (m, 4H), 2.54-2.61 (m, 2H), 2.20 (m, 4H) 2.03-2.06 (m, 1H); HPLC purity, 100.0%; SFC purity, 97.2% ee; LCMS (ESI) m/z 536.1 [M+H] + .
Example 2: Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3- morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione
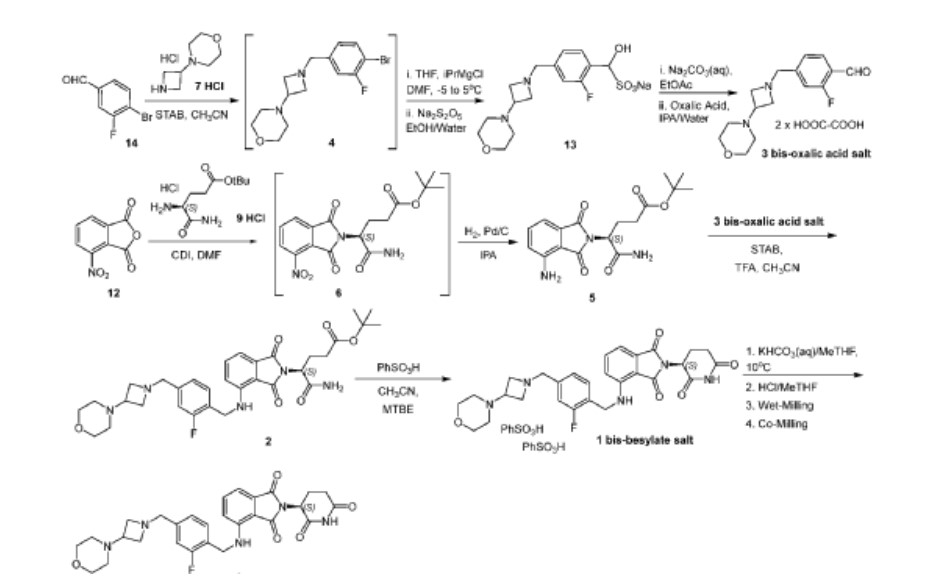
[00250] Synthesis of tert-butyl (S)-5-amino-4-(4-nitro-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 6): Ethyl acetate (245 mL, 5 V ), 3-nitrophthalic anhydride (49.1 g, 0.25 mol, 1 eq), and tert-butyl (S)-4,5-diamino-5-oxopentanoate hydrochloride (59.2 g, 0.25 mol,
1 eq) were charged into a reactor and cooled to 15-20°C. A premade solution of CDI (66.7 g,
0.41 mol, 1.5 eq) in DMF (245 mL, 5 V) was charged and the mixture was stirred at 20-25°C for
1 hour. The reaction was quenched with 15% (wt/wt) aqueous citric acid solution (10 V).
EtOAc (5 V) was added, the mixture was agitated and the phases split and separated. Tea
aqueous layer was extracted with EtOAc (5 V) and the combined organic layers were washed
twice with a 5% (wt/wt) aqueous citric acid solution (5 V each wash). The organic layer was
distilled at reduced pressure to 5 V and further continuously distilled at reduced pressure with the addition of iPrOH (10 V), maintaining a constant volume at 5 V. The final distillate was diluted to 13 V with iPrOH and used in the next step without further handling. 91% solution yield. [00251] Synthesis of tert-butyl (S)-5-amino-4-(4-amino-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 5): The solution of Compound 6 in iPrOH was charged to a hydrogenation reactor. 10% palladium on carbon (50% wet, 4.65g 5 wt%) was charged. The reaction mixture was stirred under 50-60 psi H2 at 40-50 o C for 16 hrs. The reaction mixture was filtered and the filter cake was washed three times with iPrOH (1 V each wash). The solution was distilled at reduced pressure to 5 V, cooled to ambient temperature and seeded (1 wt%). Water (20 V) was charged at 20-25 o C. The resulting slurry was cooled to 3-8 o C for 4-8 hrs. The solids were collected by filtration and washed three times with cold water (1.5 V each wash). The solids were dried at 35-45 o C under reduced pressure to give Compound 5 in 87% yield. 1 H NMR (500 MHz DMSO-d 6 ) δ (ppm): 7.52 (s, 1H), 7.43 (dd, J = 8.4, 7.0 Hz, 1H), 7.13 (s, 1H), 6.97 (ddd, J = 10.9, 7.7, 0.61 Hz, 2H), 6.43 (s, 2H), 4.49 (m, 1H), 2.33 (m, 1H), 2.17 (m, 3H), 1.32 (s, 9H); HPLC purity, 99.2%; Chiral purity, 99.9% ee; LCMS (ESI) m/z 348.2, [M+H] + , 292.2 [Mt-Bu+H] + . Residual IPA: 0.7 mol% by 1 H NMR.
[00252] Synthesis of 4-(1-(4-bromo-3-fluorobenzyl)azetidin-3-yl)morpholine (Compound 4): A mixture of 4-bromo-3-flurobenzaldehyde (Compound 14, 82 g, 396 mmol ) and 4-(azetidin-3-yl)-morpholine hydrochloride (Compound 7 HCl, 72 g, 396 mmol) in acetonitrile (820 ml) was agitated at 25±5°C for at least 3 hours. The mixture was cooled to 10±5°C and sodium triacetoxyborohydride (130 g, 594 mmol) was added in four portions while maintaining the temperature of the mixture below 30°C. The temperature of the mixture was adjusted to 25±5°C and stirred for at least 30 min until reaction completion. The mixture was transferred to a precooled (10-15°C) solution of aqueous citric acid (152 g in 400 ml water, 792 mmol) while maintaining the temperature below 30°C. Once the quenching process was complete, the mixture was concentrated to ~ 560 ml (7 volumes) while keeping the temperature at or below 45°C. The mixture was then washed with toluene (320 ml). To the aqueous phase was added THF and the pH was adjusted to above 12 with aqueous NaOH solution (320 ml, 10 N). The phases were separated, and the aqueous phase was removed. The organic phase was washed with brine and subsequently concentrated with addition of THF (~ 3L) until KF ≤ 0.10%. The mixture was filtered to remove any inorganics and the product Compound 4 was isolated as a solution in THF with 95% yield.
[00253] Synthesis of sodium (2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)phenyl) (hydroxy)methanesulfonate (Compound 13): A solution of Compound 4 (520 g, 1.58 mol) in
THF (380 ml) was cooled to −15 ± 5 °C. A solution of iPrMgCl . LiCl (1.3 M, 1823 ml, 2.37 mol) in THF was added over the course of at least 1 hour while maintaining the temperature below −10 °C. After addition was complete, the temperature of the reaction mixture was adjusted to 0 ± 5 °C and stirred for at least 1 hour. Once magnesiation was complete, the mixture was cooled to −15 ± 5 °C (target −15 °C to −20 °C) and a solution of DMF (245 ml g, 3.16 mol) in THF (260 ml) was added slowly over the course of at least 1 hour while maintaining the temperature below −10 °C. The temperature of the mixture was then adjusted to −15 ± 5 °C and agitated for at least 4 hours.
[00254] Upon reaction completion, the reaction mixture was charged into an aqueous 3 N HCl solution (2600 ml) over the course of at least 1 hour while maintaining the temperature below −5 °C. The temperature of the mixture was then adjusted to 5 ± 5 °C and agitation was stopped, letting the mixture settle for at least 15 minutes. The layers were separated. The lower aqueous layer containing the product was washed with 2-MeTHF (2600 ml). The aqueous layer was then charged with 2-MeTHF (2600 ml) and the temperature of the batch was adjusted to −10 ± 5 °C. To the cooled mixture, an aqueous 5 N NaOH (728 ml, 3.64 mol) solution was added while maintaining the temperature below −5 °C until the pH of the mixture was between 10 and 11. The temperature of the mixture was adjusted to 5 ± 5 °C and agitated for at least 15 minutes. The agitation of the mixture was stopped and the mixture allowed to settle for at least 15 minutes. The layers were separated, and the lower aqueous layer was back extracted two times with 2-MeTHF (2600 ml). The combined organic layer was washed with water (1040 mL) and the organic solution was evaporated to dryness, affording 372 g of crude Compound 3 freebase as an oil (yield 85%). 1 H NMR (DMSO-d 6 ) δ (ppm): 10.18 (s, 1H), 7.78 (t, J =7.7 Hz, 1H), 7.23-7.35 (m, 2H), 3.66 (s, 2H), 3.51 -3.60 (m, 4H), 3.26-3.47 (m, 2H), 2.72-2.97 (m, 3H), 2.12-2.32 (m, 4H).
[00255] The crude Compound 3 freebase (4.3 kg) was adsorbed onto silica gel (8.6 kg) with 100% DCM, loaded onto a 60 L column containing 12.9 kg silica gel (packed with 100% DCM), and eluted with DCM ( 86 L), followed successively by 1% MeOH/DCM (40 L), 3% MeOH/DCM (80 L) and 10% MeOH/DCM (40 L). The fractions were collected and concentrated at or below 38˚C to give Compound 3 as a purified oil (3.345 kg, yield 66%).
[00256] A portion of Compound 3 (1.0 kg, 3.59 mol) was dissolved in ethanol (16.0 L, 16
vol) at 20±5 °C and the mixture heated to 40 °C. A solution of Na2S2O5 (622.0 g, 3.27 mol; 0.91 eq) in water (2 L, 2 vol) was prepared at 20±5 °C and added to the freebase solution at 40 °C to obtain an off-white suspension. The batch was agitated and maintained at 40 °C for 2 hrs, then cooled to 20±5 °C and agitated for 1 to 2 hrs. The batch was filtered and washed with ethanol (2×2.0 L, 2×2 vol) to obtain an off-white solid. The wet cake was dried under vacuum at 40 °C for 18 hrs to afford about 1.88 kg of Compound 13.
[00257] Synthesis of 2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzaldehyde (Compound 3): Compound 13 (1.88 kg) was dissolved in ethyl acetate (15.0 L) at 20±5 °C . A 2 M Na 2 CO 3 solution (total 15.0 L used) was added to adjust the pH to 10.0. The batch was agitated for 1 to 1.5 hrs at 20±5 °C. After the reaction was complete, the phases were separated and the organic layer was washed with brine (2.0 L). The organic layer was concentrated to dryness at 35-38 °C to afford 852.0 g of Compound 3 as a colorless oil (yield 81%).
[00258] Synthesis of 2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzaldehyde bis-oxalic acid salt (Compound 3 bis-oxalic acid salt): A portion of the Compound 3 oil (187 g, 0.67 mol) was dissolved in isopropanol (1125 ml) and water (375 ml). A first portion (~30%) of this freebase mixture (480 ml) was slowly added over the course of at least 30 minutes to a solution of oxalic acid (125 g, 1.38 mol) in IPA (1125 ml)/water (375 ml) at 60 ± 5 °C. A second portion (~20%) of the freebase mixture (320 ml) was slowly added over the course of at least 30 minutes to the reaction mixture at 60 ± 5 °C. The reaction mixture was agitated at 60 ± 5 °C for at least 90 minutes. A third portion (~25%) of the freebase mixture (~ 400 ml) was slowly added over the course of at least 30 minutes to the reaction mixture at 60 ± 5 °C and the reaction mixture was agitated at 60 ± 5 °C for at least 90 minutes. The remaining freebase solution (400 ml) was slowly added over the course of at least 30 minutes to the reaction mixture at 60 ± 5 °C and the reaction mixture was agitated at 60 ± 5 °C for at least 90 minutes. The temperature of the mixture was adjusted to 20 ± 5 °C (target 20 °C) over the course of at least 1 hour and the mixture was agitated for at least 16 hours at 20 ± 5 °C and then filtered. The cake was washed three times with IPA (2 x 375 ml) and dried in the drying oven at ≤ 40 °C with a slow bleed of nitrogen to afford 261 g of Compound 3 bis-oxalic acid salt (yield 85%). 1 H NMR (DMSO-d 6 ) δ (ppm): 10.21 (s, 1H), 7.87 (t, J = 7.6 Hz, 1H), 7.42-7.56 (m, 2H), 4.31 (s, 2H), 3.89 -4.03 (m, 2H), 3.75-3.89 (m, 2H), 3.60 (br t, J = 4.3 Hz, 4H), 3.26 (br t, J = 6.9 Hz, 1H), 2.37 (br s, 4H) .
[00259] Synthesis of tert-butyl (S)-5-amino-4-(4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)-1,3- dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 2):
Acetonitrile (6.8 L, 8.0 X Vol) was added to a 30 L jacketed cylindrical reactor. Compound 5 (0.845 kg, 1.00 X Wt) and Compound 3 bis-oxalic acid salt (1.35 kg, 1.60 The contents of the reactor were equilibrated with agitation at 20 ± 5 °C. Trifluoroacetic acid (0.19 L, 0.22 X Vol) was added dropwise, maintaining the batch temperature at 20 ± 5 °C. The reaction mixture was stirred at 20 ± 5 °C for no less than 5 minutes and then sodium triacetoxyborohydride (0.13 kg, 015 X Wt) was added as a solid, maintaining the batch temperature at 20 ± 5 °C. The process of adding trifluoroacetic acid and then sodium triacetoxyborohydride was repeated an additional 5 times. After the last addition, the reaction mixture was sampled to determine the reaction progress. The reaction was held at 20 ± 5 °C overnight. The reaction mixture was then quenched with water (3.4 L, 4.0 X Vol), maintaining the batch temperature at 20 ± 5 °C. The mixture was then stirred at 20 ± 5 °C for no less than 30 minutes and the resulting slurry filtered through a 3 L sintered glass filter, directing the filtrates to clean containers. The reactor was rinsed with acetonitrile (0.4 L, 0.5 X Vol) and the rinse passed through the contents of the 3 L sintered glass filter, directing the filtrate to the containers containing the main batch. The contents of the containers were concentrated to ~5 X Vol under reduced pressure at a bath temperature of no more than 30 °C. The residue was transferred to a clean reactor, was rinsing with 2-MeTHF (2.5 L, 3.0 X Vol) to complete the transfer. Additional 2-MeTHF (10.1 L, 12.0 X Vol) was added to the reactor, followed by water (3.4 L, 4.0 X Vol). The mixture was agitated for no less than 15 minutes at 20 ± 5 °C, then allowed to settle for no less than 10 minutes at 20 ± 5 °C before transferring the bottom aqueous layer to new containers. An aqueous sodium bicarbonate solution (5.3 L, 6.3 X Vol, 9% wt/wt) was added to the reactor with stirring over 30 minutes, maintaining batch temperature no more than 25 °C. The mixture was agitated for no more than 15 minutes at 20 ± 5 °C, then allowed to settle for no less than 10 minutes at 20 ± 5 °C before the bottom aqueous layer was transferred to new containers. The aqueous sodium bicarbonate wash was repeated an additional 2 times to reach a pH of about 6.6 for the spent aqueous layer. A saturated aqueous solution of NaCl(0.85 L, 1.0 X Vol) was then added to reactor with agitation. The mixture was agitated for no less than 15 minutes at 20 ± 5 °C, then allowed to settle for no less than 10 minutes before the bottom aqueous layer was transferred to new containers. The remaining organics were concentrated under reduced pressure to a batch volume of ~5 X Vol at a bath temperature of about 40 °C. Acetonitrile (5.1 L, 6.0 X Vol) was added to the residual volume and the resulting solution concentrated to a batch volume of ~ 5 X Vol under reduced pressure at bath temperature of about 40 °C. The process of adding acetonitrile and concentrating under vacuum was repeated two more times to reach the distillation endpoint with a water content of about 1%. The acetonitrile solution was transferred to a clean container along with two 1.7 L (2.0 X Vol) rinses and held at 5 °C overnight. The acetonitrile solution was then filtered through a 3 L sintered glass filter, followed by a 1.7 L (2.0 X Vol) acetonitrile rinse, directing the filtrates to a clean container. The filtrate was transferred to a clean reactor and the container rinsed twice with 1.7 L (2.0 X Vol) of acetonitrile to complete the transfer. Enough acetonitrile (roughly 0.6 L) was added to adjust the total volume in the reactor to about 14 L. A solution assay of the contents of the reactor was obtained to calculate the amount of Compound 2 present for use in the next step (result = 1.3 kg = 1.00 X Wt for remainder of process).
[00260] Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione bis-besylate salt (Compound 1 bis-besylate salt): The solution of Compound 2 in acetonitrile from the previous step was diluted with acetonitrile (roughly 2 L) such that the total volume in the reactor was about 16 L. The solution was cooled with stirring to 10 ± 5 °C and held within that range for 96 hours. Benzenesulfonic acid (1.86 kg, 1.43 X Wt) was added while sparging the reaction mixture with nitrogen gas and maintaining the batch temperature at 10 ± 10 °C. The temperature of the reactor was then adjusted to 20 ± 5 °C and the mixture stirred at that temperature for 60 minutes. The total volume of reaction mixture was adjusted back to 16 L to account for solvent lost during sparging by the addition of acetonitrile (roughly 0.4 L). The reaction mixture was then heated to 55 ± 5 °C over the course of about 30 minutes and held in that range for 15 to 16 hours for reaction completion. The mixture was then cooled to 50 ± 5 °C and MTBE (3.9 L, 3.0 X Vol) was added, maintaining the batch temperature at 50 ± 5 °C. The mixture was allowed to stir at 50 ± 5 °C for about 1.5 hours to establish a self-seeded slurry. Additional MTBE (3.9 L, 3.0 X Vol) was added to the reactor over the course of about 1.75 hours at 50 ± 5 °C. The slurry was cooled to 20 ± 5 °C over the course of about 1.75 hours and held in that temperature range overnight. The slurry was filtered using a Buchner funnel. The reactor was rinsed twice with
MTBE (3.9 L each, 3.0 X Vol) and the rinse was used to wash the solids in the Buchner funnel. The solids were dried on drying trays for about 23 hours at 40 °C under reduced pressure (15-150 mbar), yielding 1.62 kg (77.9%) of Compound 1 bis-besylate salt.
[00261] Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione hydrochloride salt (Compound 1 HCl): A suspension of Compound 1 bis-besylate salt (120 g, 1 equiv.) in 2-MeTHF (25 L/kg) was added to a reactor and agitated at 10 °C. A solution of KHCO 3 (32.5 g, 2.4 equiv) in water (1.8 L, 6 L/kg) was added to the slurry over the course of 40 minutes. The mixture was stirred for an additional 30 minutes. The batch was then allowed to settle, at which point the aqueous (bottom) layer was separated and discarded. An aqueous solution of NaCl (5%, 5 L/kg, 575 ml) was added to the organic layer and the mixture was agitated for 10 minutes, after which point the temperature was raised to 20 °C. The batch was allowed to settle, at which point the aqueous (bottom) layer was discarded. The brine was repeated a second time.
Additional 2-MeTHF (500 ml) was added to dilute the organic layer, resulting in a concentration of about 20 mg product per ml. A solution of HCl (total 0.98 eq.) in 2-MeTHF was prepared and a portion (20% of total, corresponding to ~0.2 eq.) then added to the reaction mixture over the course of about 10 min. Seeds of Compound 1 hydrochloride (~5% wt) were added, but did not dissolve. The batch was held under vigorous agitation for one hour. To the slurry, the remaining portion of the HCl solution (~0.78 eq.) was added over the course of 3 hours at a constant rate. Vigorous agitation was maintained. After addition was complete, the batch was held for one hour, after which the batch was filtered, washed three times with 3 L/kg of 2-MeTHF . The filter cake was placed in a vacuum oven at 22 °C for 12 hours, at which point the temperature was raised to 40 °C. Dry cake of Compound 1 hydrochloride (58g, 75% yield) was obtained and packaged. Achiral HPLC purity: 98.91%; chiral HPLC purity: 99.68%.
Example 4: Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3- morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione
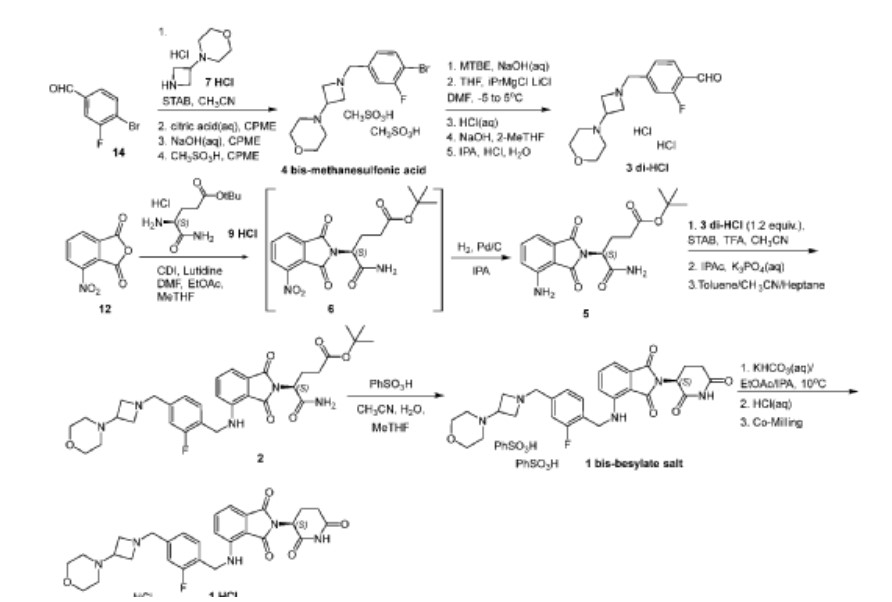
[00264] Synthesis of tert-butyl (S)-5-amino-4-(4-nitro-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 6): To a solution of 3-nitrophthalic anhydride (Compound 12,
35.15 g, 176.6 mmol, 1.00 eq) in ethyl acetate (350 mL) was added tert-butyl (4S)-4,5-diamino-5-oxo-pentanoate hydrochloride (Compound 9 HCl, 43.22 g, 181.1 mmol, 1.025 eq) ), DMF (70
mL) and 2-MeTHF (110 mL) at 25°C. 2,6-Lutidine (23.4 mL, 201 mmol, 1.14 eq) was added
slowly to maintain the temperature at or below 25°C. The mixture was aged at 25°C for 1 hour before being cooled to 5°C. CDI (4.17 g, 25.7 mmol, 0.146 eq) was added and stirred until the temperature returned to 5°C. Another portion of CDI (4.62 g, 28.5 mmol, 0.161 eq) was added and stirred until temperature returned to 5°C. CDI (8.87 g, 54.7 mmol, 0.310 eq) was added and stirred until the temperature returned to 5°C. CDI (8.91 g, 54.9 mmol, 0.311 eq) was added and stirred until the temperature returned to 5°C. The mixture was warmed to 20°C and CDI (16.4 g, 101.1 mmol, 0.573 eq) was added, and the mixture was aged at 20°C for 16 hours. The mixture was cooled to 5°C and a solution of 30 wt% citric acid and 5 wt% NaCl (350 mL) was added slowly while maintaining the temperature. The mixture was warmed to 20°C and aged for 30 minutes. The phases were split and separated. The organic phase was diluted with EtOAc (175 mL) and washed with a solution of 5 wt% citric acid (175 mL), and concentrated by distillation (75 torr, 50°C) to a volume of 175 mL EtOAc. The solvent was changed to iPrOH by constant volume distillation (75 torr, 50°C) with 350 mL iPrOH to a final volume of 175 mL. The distillate was diluted with 200 mL iPrOH to afford Compound 6 as a solution for use in the next step. 1 H NMR (500 MHz, CDCl 3 ) δ (ppm): 8.18 – 8.13 (m, 2H), 7.96 (t, J = 7.8 Hz, 1H), 6.34 (s, 1H), 5.59 (s, 1H), 4.90 (dd, J = 10.1, 4.6 Hz, 1H), 2.61 (ddt, J = 14.6, 10.1, 6.1 Hz, 1H), 2.49 (ddt, J = 14.2, 8.7, 5.2 Hz, 1H), 2.44 – 2.29 ( m, 2H), 1.44 (s, 9H).
[00265] Synthesis of tert-butyl (S)-5-amino-4-(4-amino-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 5): To a solution of Compound 6 in iPrOH (375 mL) was added 5% palladium on carbon (1.23 g, 3.5 wt%, wet). The mixture was purged with nitrogen five times and with hydrogen three times. The mixture was pressurized with hydrogen (50 psi) and aged at 50°C for 16 hours. The mixture was cooled to room temperature and purged with nitrogen three times, filtered to remove catalyst, and the filter cake was washed with iPrOH (20mL) three times. The filtrate was concentrated to 200 mL, seeded (0.454 g, 1.3 wt%) at 22 °C, and aged for 45 minutes. Water (1325 mL) was added over 3 hours at 22°C. After the addition of water, the mixture was cooled to 8°C over 2 hours and aged for 1 hour at 8°C. The slurry was filtered, and the cake was rinsed with cold water (200 mL) three times and dried under vacuum at 50°C to yield Compound 5 as a yellow solid (47.97 g, 80.6% yield, 99.62% LC purity, 103% 1 H NMR potency). 1 H NMR (500 MHz, CDCl 3 ) δ (ppm): 7.46 (dd, J = 8.3, 7.0 Hz, 1H), 7.19 (d, J = 7.2 Hz, 1H), 6.89 (d, J = 8.3 Hz, 1H), 6.28 (s, 1H), 5.41 (s, 1H), 5.28 (s, 2H), 4.83 (dd, J = 9.3, 6.0 Hz, 1H), 2.52 (p, J = 7.0 Hz, 2H), 2.36 – 2.29 (m, 2H), 1.44 (s, 9H). 13 C NMR (126 MHz, CDCl 3 ) δ (ppm): 171.80, 171.12, 169.64, 168.27, 145.70, 135.50, 132.20, 121.43, 112.98, 80.99, 53.04, 32.23, 28.02 , 24.36. LCMS (ESI): m/z 291.9 [M+H – tBu]
[00266] Synthesis of 4-(1-(4-bromo-3-fluorobenzyl)azetidin-3-yl)morpholine bis-methanesulfonic acid salt (Compound 4 bis-methanesulfonic acid salt): A mixture of 4-bromo-3- fluorobenzaldehyde (Compound 14, 102 g, 493 mmol) and 4-(azetidin-3-yl)morpholine hydrochloride (Compound 7 HCl, 90 g, 493 mmol) in acetonitrile (1000 ml) was agitated at a temperature of about 20 to 25 °C for 2 to 3 hours. The slurry was cooled to temperature of about 10 to 15 °C and sodium triacetoxyborohydride (STAB, 162 g, 739 mmol) was added in 4 portions over the course of about 45 minutes while maintaining the batch temperature at no more than 30 °C. The slurry was stirred at a temperature of about 20 to 25 °C for at least 30 minutes and then quenched by an aqueous citric acid solution (191 g, 986 mmol in 500 ml of water) at a temperature of about 40 to 45 °C over the course of 2 hours. Upon completion of the quenching process, the batch volume was reduced by vacuum distillation to about 700 ml at a temperature of no more than 45 °C. Cyclopentylmethylether (CPME, 400 ml) was added to the aqueous solution to afford a final volume of about 1100 ml. The pH was adjusted to about 8 to 9 by addition of an aqueous solution of 10 N NaOH (added volume about 430 ml). The phases were separated, and the aqueous phase discarded. The organic phase was washed with brine (100 ml) twice such that the pH was no more than 8 and the volume was adjusted to about 1000 ml with addition of extra CPME. The batch was distilled at constant volume under reduced pressure with addition of CPME until KF was no more than 0.15%. CPME was added (if needed) to adjust the batch to a volume of 1000 ml at the end of distillation. The dry CPME solution was seeded (500 to 750 mg) at ambient temperature. The seeded, dry CPME slurry was heated to a temperature of 50 to 60 °C and then charged with methanesulfonic acid in 200 ml of CPME over the course of 4 to 5 hours. The slurry was then cooled to 20 °C over the course of 4 to 5 hours and kept at 20 °C for 3 to 4 hours, filtered, rinsed with CPME and dried in a vacuum oven at 35 to 40 °C over 16 hours to give Compound 4 bis-methanesulfonic acid salt as a white solid. 1 H NMR (500 MHz DMSO-d 6 ) δ (ppm): 10.62 (br s, 1-2H), 7.85 (t, J = 7.8 Hz, 1H), 7.58 (dd, J = 9.5 Hz, 1.9 Hz, 1H), 7.34 (dd, J = 8.2 Hz, 1.8 Hz, 1H), 4.55-4.24 (m, 7H), 3.84 (br s, 4H), 3.14 (m, 4H); HPLC purity, 99.8%, LCMS (ESI) m/z 329.1 /331.1[M/M+2] + . XRPD pattern of the product is shown in FIG.10. DSC thermogram of the product is shown in FIG.11.
[00267] Preparation of 4-(1-(4-bromo-3-fluorobenzyl)azetidin-3-yl)morpholine (Compound 4): A slurry of Compound 4 bis-methanesulfonic acid salt (70 g, 134 mmol) in t -butyl methyl ether was cooled to 10±5°C. An aqueous solution of NaOH (2 N, 201 ml, 403 mmol) was added over the course of at least 30 minutes while maintaining the batch temperature at about 15 °C. After the addition of NaOH , the batch temperature was raised to 20±5°C and agitated over the course of about 20 minutes. The organic layer was separated and washed with water (210 ml) three times. The organic layer was subsequently concentrated with addition of THF (~ 1.05L) until KF ≤ 0.10%. The product Compound 4 was isolated as a solution in THF
with 95% solution yield.
[00268] Preparation of 2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzaldehyde dihydrochloride (Compound 3 di-HCl): A solution of Compound 4 (44 g, 134 mmol) in THF (total volume ~ 350 ml) was then cooled to −20 ± 5 °C. A solution of iPrMgCl . LiCl (1.3 M, 176 ml, 228 mmol) in THF was added over the course of half an hour while maintaining the temperature below −10 °C. After the addition was complete, the batch was stirred at −20 ± 5 °C for 16 to 22 hours. DMF (21 ml, 268 mmol) was then added slowly over the course of 30 minutes while maintaining the batch temperature no more than -15 °C. The batch was stirred at −20 ± 5 °C for 6 to 24 hours. 2-MeTHF (350 ml) was then added to the batch over the course of 30 minutes, followed by slow addition of 3 N HCl (235 ml, 704 mmol) while keeping the batch temperature no more than -10 °C. After the addition of aqueous HCl, the batch was warmed to 0 ± 5 °C and 2 N aqueous NaOH (154 ml, 309 mmol) was added slowly to adjust the solution pH to about 8 to 9. The batch was stirred for about 30 minutes and then warmed to 20 ± 5 °C. The organic layer was separated and washed with 15% aqueous NaCl (3 x 140 ml). The organic layer was subsequently concentrated with addition of 2-MeTHF until KF ≤ 0.10%.
[00269] A portion of the free base of 2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzaldehyde (37.4 g, 134 mmol) so obtained was dissolved in 2-MeTHF (total ~ 420 ml) , to which isopropanol (420 ml) and water (21 ml) were added at 20 ± 5 °C. The batch was then heated to 50 ± 5 °C and a solution of HCl in IPA (5 to 6 N, 28 ml, half of total HCl volume) was added over the course of 1 hour. The batch was seeded with 2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzaldehyde dihydrochloride (700 mg) and aged for 1 hour. The remaining HCl (28 ml) was then added over the course of 1 hour. The batch was agitated at 50 ± 5 °C for 4 hours and then cooled to 20 ± 5 °C for 8 hours. The slurry was filtered, washed with IPA (210 ml), and the filter cake dried under vacuum at 50 ± 5 °C to afford Compound 3 dihydrochloride salt (36 g, yield 75%). 1 H NMR (DMSO-d 6 ) δ (ppm): 12.32-12.55 (m, 1H), 10.23 (s, 1H), 7.93 (t, J =7.6 Hz, 1H), 7.66 (d, J = 10.5 Hz , 1H), 7.58 (d, J = 7.9 Hz, 1H), 4.80 (br s, 2H), 4.48-4.70 (m, 2H), 4.30 (br s, 4H), 3.78-4.00 (m, 5H), 2.93-3.15 (m, 2H). Two polymorphic forms were obtained. XRPD pattern and DSC thermogram of Form A (anhydrous) are shown in FIG.5 and FIG.6, respectively. XRPD pattern, TGA thermogram and DSC thermogram of Form B (hydrate) are shown in FIG.7, FIG.8, and FIG.9, respectively.
[00270] Synthesis of tert-butyl (S)-5-amino-4-(4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)-1,3- dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 2): A mixture of Compound 5 (12 g, 34.5 mmol, 1.0 eq) and Compound 3 dihydrochloride (14.56 g, 41.5 mmol, 1.2 eq) in MeCN (96 ml) was cooled to 0-5 °C. Trifluoroacetic acid (TFA, 2.0 ml, 26 mmol, 0.75 eq) was added, followed by sodium triacetoxyborohydride (STAB, 2.75 g, 12.95 mmol, 0.375 eq) while maintaining the internal temperature below 10 °C. The addition of TFA and STAB was repeated three additional times. After a total of four additions of TFA and STAB, the reaction was aged at 0-5°C for 1 hour. A 10% brine solution (108 ml) was then added to the reaction mixture over the course of 1 hour and partitioned with IPAc (96 ml). The mixture was warmed to 20-25 °C and aged for 30 minutes. The layers were then separated and the organic layer was washed with 2.0 M K3PO4 (114 ml). The pH of the spent aqueous layer should have a pH of about 8.5 – 9.0. The layers were separated again and the organic phase was washed with 8.5% NaHCO 3 (2 x 60 ml), with 30 minutes between each wash, followed by 24% brine (60 ml). The organic fraction was distilled to 72 ml at an internal temperature near 50°C. Toluene (72 ml) was added to bring the volume to 144 ml and distillation continued at constant volume at 50°C with feed and bleed until water content < 0.1. The mixture was heated to 50°C and acetonitrile (48 ml) was added, followed by slow addition of heptane (144 ml) while maintaining the internal temperature above 45 °C. The reaction was held at 50 °C for 2 hours. Once complete, the reaction was slowly cooled to 20-25°C over the course of 4 hours and held at 20-25°C overnight (16 hour). The yellow slurry was then filtered and the yellow cake displacement washed with 1:3:3 mixture of acetonitrile/heptane/toluene (3 x 48 ml). The final cake was then dried under reduced pressure at 50 °C under nitrogen to provide Compound 2 (87.7% isolated molar yield) with >99.0% LCAP. HPLC purity, 99.85%; Chiral purity, >99.9% ee. 1 H NMR (DMSO-d6, 500 MHz) δ (ppm) 7.55 (s, 1H), 7.51 (dd, J = 7.2, 8.4 Hz, 1H), 7.32 (t, J = 7.9 Hz, 1H), 7.16 ( s, 1H), 7.0-7.1 (m, 5H), 4.57 (d, J = 6.3 Hz, 2H), 4.5-4.5 (m, 1H), 3.5-3.6 (m, 6H), 3.3-3.4 (m, 3H), 2.8-2.9 (m, 3H), 2.3-2.4 (m, 1H), 2.1-2.3 (m, 7H), 1.31 (s, 9H); LCMS m/z 610.3 [M+H] + .
[00271] Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione bis-besylate salt (Compound 1 bis-besylate): To a suspension of Compound 2 (130 g, 1.0 equiv.) in MeCN (1.56 L, 12 L/kg) agitated at 55 °C was added a solution of benzene sulfonic acid (185 g, 5.5
equiv.) in MeCN (0.39 L, 3 L/kg) and water (0.01 L, 2.0 equiv.). The mixture was stirred at 55 °C for 16 hours. After the reaction age, crystalline seeds (1.3 g, 1 wt%) of bis-besylate salt of Compound 1 were charged into the batch, resulting in formation of a yellow slurry. The slurry was then cooled to 20 °C over the course of 90 minutes. 2-MeTHF (1.3 L, 10 L/kg) was added to the batch slowly over 2 hours at 20 °C. The batch was agitated for an additional 4 hours at 20°C. The yellow slurry was then filtered and the yellow cake re-slurried with MeTHF (1.3 L, 10 L/kg) followed by a displacement MeTHF (0.65 L, 5 L/kg) wash. The final cake was then dried under reduced pressure at 50 °C under nitrogen to give Compound 1 bis-besylate salt (160 g, 88.4% yield). HPLC purity: 98.39%; chiral HPLC purity: 100%. XRPD pattern, TGA thermogram and DSC thermogram of the product are shown in FIG.1, FIG.2, and FIG.3, respectively.
[00272] Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione hydrochloride salt (Compound 1 HCl): A suspension of Compound 1 bis-besylate salt (300 g, 1 equiv.) in EtOAc (4.68 L, 15.6 L/kg) and 2-propanol (0.12 L, 0.4 L/kg) was agitated at 15 °C. To the suspension was added a solution of KHCO 3 (82.4 g, 2.5 equiv) in water (1.8 L, 6 L/kg) over 30 minutes. The mixture was heated to 20 °C over 30-60 minutes and then agitated for 30 minutes. The batch was allowed to settle for 30 minutes, at which point the aqueous (bottom) layer was discarded. To the rich organic layer was added water (1.2 L, 4 L/kg) and the reactor contents were agitated for 30 minutes. The batch was allowed to settle for 30 minutes, at which point the aqueous (bottom) layer was discarded. To the rich organic stream was added 2-propanol (2.375 L, 7.9 L/kg) and the stream was then filtered. Water was added to the filtrate to adjust the water content to 8≤KF≤8.2. To the above agitated solution at 20 °C was added 0.2 N HCl (38 mL, 0.025 equiv prepared in EtOAC/IPA 2:1, v/v with 8wt% water) over 10 minutes. To the mixture was added crystalline seeds of Compound 1 hydrochloride salt (1.6 g, 0.5wt%) and the contents of the reactor were agitated at 20 °C for 30 minutes. To the suspension was added 0.2 N HCl (1.44 L, 0.945 equiv. prepared in EtOAC/IPA 2:1, v/v with 8 wt% water) over 4.5 hours. The slurry was agitated for 14 hours, then filtered and washed with EtOAC/IPA (750 mL, 2.5 L/kg, 2:1 v/v with 8 wt% water) followed by IPA (750 mL, 2.5 L/kg). The solids were dried under vacuum at 40 °C to afford Compound 1 hydrochloride salt (170 g, 90% yield). Achiral HPLC purity: 99.91%; chiral HPLC purity: 99.58%. XRPD analysis (FIG.4) confirmed the product (a)


AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
//////////Golcadomide, CC 99282, CC 1007548, WHO 12305
C1CC(=O)NC(=O)C1N2C(=O)C3=C(C2=O)C(=CC=C3)NCC4=C(C=C(C=C4)CN5CC(C5)N6CCOCC6)F
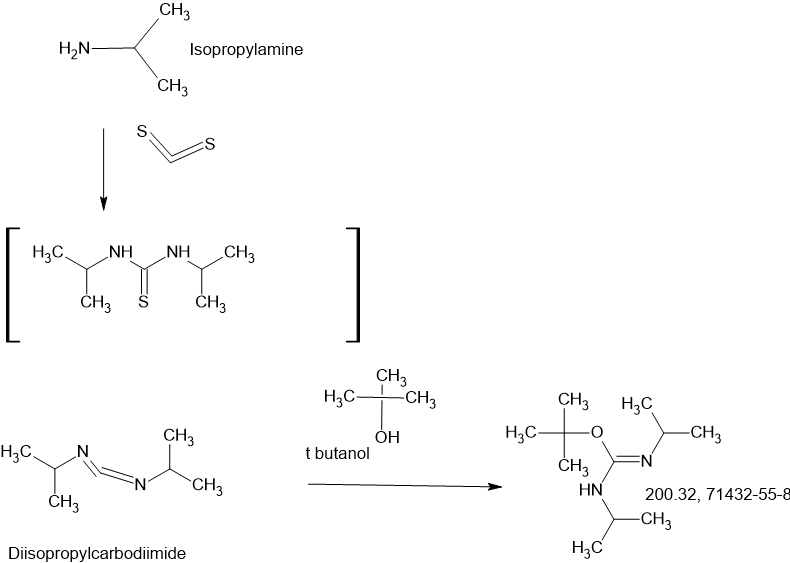
SIDECHAIN
MAIN