















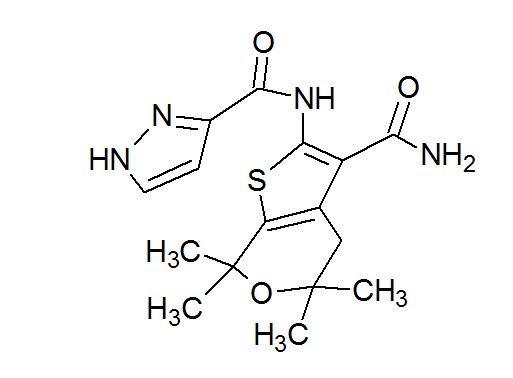
GLGP 1837
CAS 1654725-02-6
MF C16 H20 N4 O3 S, MW 348.42
For cystic fibrosis treatment
| N-(3-carbamoyl-5,5,7,7-tetramethyl-4H-thieno[2,3-c]pyran-2-yl)-1H-pyrazole-5-carboxamide |
1H-Pyrazole-3-carboxamide, N-[3-(aminocarbonyl)-4,7-dihydro-5,5,7,7-tetramethyl-5H-thieno[2,3-c]pyran-2-yl]-
| Inventors | Der Plas Steven Emiel Van, Sébastien Laurent Xavier MARTINA, Sébastien Jean-Jacques Cédric DROPSIT-MONTOVERT, Martin James Inglis Andrews, Hans KELGTERMANS |
| Applicant | Galapagos Nv |
![]()
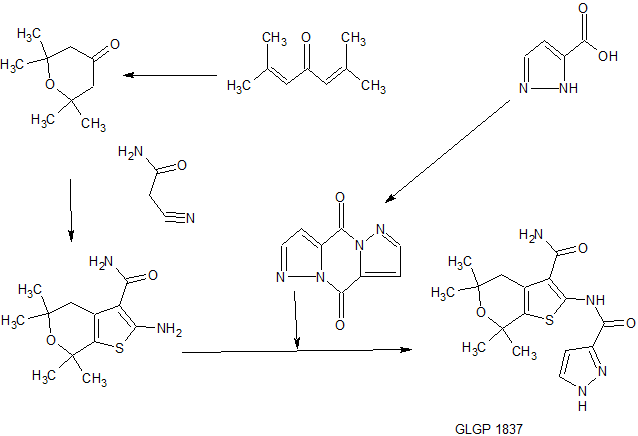
SYNTHESIS

GLGP 1837
ABC transporters are a family of homologous membrane transporter proteins regulating the transport of a wide variety of pharmacological agents (for example drugs, xenobiotics, anions, etc…) that bind and use cellular adenosine triphosphate (ATP) for their specific activities. Some of these transporters were found to defend malignant cancer cells against chemotherapeutic agents, acting as multidrug resistance proteins (like the MDRl-P glycoprotein, or the multidrug resistance protein, MRP 1). So far, 48 ABC transporters, grouped into 7 families based on their sequence identity and function, have been identified.
ABC transporters provide protection against harmful environmental compounds by regulating a variety of important physiological roles within the body, and therefore represent important potential drug targets for the treatment of diseases associated with transporter defects, outwards cell drug transport, and other diseases in which modulation of ABC transporter activity may be beneficial.
The cAMP/ATP -mediated anion channel, CFTR, is one member of the ABC transporter family commonly associated with diseases, which is expressed in a variety of cells types, including absorptive and secretory epithelia cells, where it regulates anion flux across the membrane, as well as the activity of other ion channels and proteins. The activity of CFTR in epithelial cells is essential for the maintenance of electrolyte transport throughout the body, including respiratory and digestive tissue. (Quinton, 1990)
The gene encoding CFTR has been identified and sequenced (Kerem et al., 1989). CFTR comprises about 1480 amino acids that encode a protein made up of a tandem repeat of transmembrane domains, each containing six transmembrane helices and a nucleotide binding domain. The pair of
transmembrane domains is linked by a large, polar, regulatory (R)-domain with multiple phosphorylation sites that regulate channel activity and cellular trafficking.
Cystic fibrosis is caused by a defect in this gene which induces mutations in CFTR. Cystic fibrosis is the most common fatal genetic disease in humans, and affects -0.04% of white individuals(Bobadilla et al., 2002), for example, in the United States, about one in every 2,500 infants is affected, and up to 10 million people carry a single copy of the defective gene without apparent ill effects; moreover subjects bearing a single copy of the gene exhibit increased resistance to cholera and to dehydration resulting from diarrhea. This effect might explain the relatively high frequency of the CF gene within the population.
In contrast, individuals with two copies of the CF associated gene suffer from the debilitating and fatal effects of CF, including chronic lung infections.
In cystic fibrosis patients, mutations in endogenous respiratory epithelial CFTR fails to confer chloride and bicarbonate permeability to epithelial cells in lung and other tissues, thus leading to reduced apical anion secretion and disruptions of the ion and fluid transport. This decrease in anion transport causes an enhanced mucus and pathogenic agent accumulation in the lung triggering microbial infections that ultimately cause death in CF patients.
Beyond respiratory disease, CF patients also suffer from gastrointestinal problems and pancreatic insufficiency that result in death if left untreated. Furthermore, female subjects with cystic fibrosis suffer from decreased fertility, whilst males with are infertile.
A variety of disease causing mutations has been identified through sequence analysis of the CFTR gene of CF chromosomes (Kerem et al., 1989). AF508-CFTR, the most common CF mutation (present in at least 1 allele in~90 % of CF patients) and occurring in approximately 70% of the cases of cystic fibrosis, contains a single amino acid deletion of phenylalanine 508. This deletion prevents the nascent protein from folding correctly, which protein in turn cannot exit the endoplasmic reticulum (ER) and traffic to the plasma membrane, and then is rapidly degraded. As a result, the number of channels present in the membrane is far less than in cells expressing wild-type CFTR. In addition to impaired trafficking, the mutation results in defective channel gating. Indeed, even if AF508-CFTR is allowed to reach the cell plasma membrane by low-temperature (27°C) rescue where it can function as a cAMP-activated chloride channel, its activity is decreased significantly compared with WT-CFTR (Pasyk and Foskett, 1995).
Other mutations with lower incidence have also been identified that alter the channel regulation or the channel conductance. In case of the channel regulation mutants, the mutated protein is properly trafficked and localized to the plasma membrane but either cannot be activated or cannot function as a chloride channel (e.g. missense mutations located within the nucleotide binding domains), examples of these mutations are G551D, G178R, G1349D. Mutations affecting chloride conductance have a CFTR protein that is correctly trafficked to the cell membrane but that generates reduced chloride- flow (e.g. missense mutations located within the membrane-spanning domain), examples of these mutations are Rl 17H, R334W.
In addition to cystic fibrosis, CFTR activity modulation may be beneficial for other diseases not directly caused by mutations in CFTR, such as, for example, chronic obstructive pulmonary disease (COPD), dry eye disease, and Sjogren’s Syndrome.
[0014] COPD is characterized by a progressive and non-reversible airflow limitation, which is due to mucus hypersecretion, bronchiolitis, and emphysema. A potential treatment of mucus hypersecretion and impaired mucociliary clearance that is common in COPD could consist in using activators of mutant or wild-type CFTR. In particular, the anion secretion increase across CFTR may facilitate fluid transport into the airway surface liquid to hydrate the mucus and optimize periciliary fluid viscosity. The resulting enhanced mucociliary clearance would help in reducing the symptoms associated with COPD.
[0015] Dry eye disease is characterized by a decrease in tear production and abnormal tear film lipid, protein and mucin profiles. Many factors may cause dry eye disease, some of which include age, arthritis, Lasik eye surgery, chemical/thermal burns, medications, allergies, and diseases, such as cystic fibrosis and Sjogrens’s syndrome. Increasing anion secretion via CFTR could enhance fluid transport from the corneal endothelial cells and secretory glands surrounding the eye, and eventually improve corneal hydration, thus helping to alleviate dry eye disease associated symptoms. Sjogrens’s syndrome is an autoimmune disease where the immune system harms moisture-producing glands throughout the body, including the eye, mouth, skin, respiratory tissue, liver, vagina, and gut. The ensuing symptoms, include, dry eye, mouth, and vagina, as well as lung disease. Sjogrens’s syndrome is also associated with rheumatoid arthritis, systemic lupus, systemic sclerosis, and polymypositis/dermatomyositis. The cause of the disease is believed to lie in defective protein trafficking, for which treatment options are limited. As a consequence, modulation of CFTR activity may help hydrating the various organs and help to elevate the associated symptoms.
In addition to CF, the defective protein trafficking induced by the AF508-CFTR has been shown to be the underlying basis for a wide range of other diseases, in particular diseases where the defective functioning of the endoplasmic reticulum (ER) may either prevent the CFTR protein to exit the cell, and/or the misfolded protein is degraded (Morello et al., 2000; Shastry, 2003; Zhang et al., 2012).
[0017] A number of genetic diseases are associated with a defective ER processing equivalent to the defect observed with CFTR in CF such as glycanosis CDG type 1, hereditary emphysema (α-1-antitrypsin (PiZ variant)), congenital hyperthyroidism, osteogenesis imperfecta (Type I, II, or IV procollagen), hereditary hypofibrinogenemia (fibrinogen), ACT deficiency (α-1-antichymotrypsin), diabetes insipidus (DI), neurophyseal DI (vasopvessin hormoneN2 -receptor), neprogenic DI (aquaporin II), Charcot-Marie Tooth syndrome (peripheral myelin protein 22), Perlizaeus-Merzbacher disease, neurodegenerative diseases such as Alzheimer’s disease (APP and presenilins), Parkinson’s disease, amyotrophic lateral sclerosis, progressive supranuclear plasy, Pick’s disease, several polyglutamine neurological disorders such as Huntington’s disease, spinocerebullar ataxia type I, spinal and bulbar muscular atrophy,
dentatorubal pallidoluysian, and myotonic dystrophy, as well as spongiform encephalopathies, such as hereditary Creutzfeldt-Jakob disease (prion protein processing defect), Fabry disease (lysosomal a-galactosidase A), Straussler-Scheinker syndrome, chronic obstructive pulmonary disease (COPD), dry eye disease, and Sjogren’s Syndrome.
In addition to up-regulation of the activity of CFTR, anion secretion reduction by CFTR modulators may be beneficial for the treatment of secretory diarrheas, in which epithelial water transport is dramatically increased as a result of secretagogue activated chloride transport. The mechanism involves elevation of cAMP and stimulation of CFTR.
[0019] Regardless of the cause, excessive chloride transport is seen in all diarrheas, and results in dehydration, acidosis, impaired growth and death. Acute and chronic diarrheas remain a major medical problem worldwide, and are a significant factor in malnutrition, leading to death in children of less than five years old (5,000,000 deaths/year). Furthermore, in patients with chronic inflammatory bowel disease (IBD) and/or acquired immunodeficiency syndrome (AIDS), diarrhea is a dangerous condition
GLGP 1837
PATENT
Scheme 1: synthesis of the core and subsequent amide coupling

O
1 M HCI
amide coupling
HO Λ R-i

Example 2. Synthesis of intermediates
Intermediate 2: 2,2, 6,6-tetramethyltetrahydro-4H-pyran-4-one

Phorone or 2,6-dimethyl-2,5-heptadien-4-one (1 eq) is mixed with an aqueous 1 M HCI solution and the obtained emulsion is stirred at 40°C for 6 days. The water phase is extracted with DCM, and the organic phase is concentrated and purified by distillation to afford the desired product.
Alternative synthesis of Intermediate 2
[00208] A 20 L reactor is charged with aqueous 6M HCI and is warmed up to 30 °C. Molten Phorone is added while stirring vigorously at 40°C for up to 3 h until completion. The resulting solution is then cooled to 30°C and extracted with 4 x 1 L DCM. The combined organic phases are washed with saturated NaHC03 solution (400 niL) and are dried over Na2S04. The resulting crude misture is then concentrated under vacuo, and finally purified by distillation.
Intermediate 3: 2-Amino-5,5, 7, 7-tetramethyl-4, 7-dihydro-5H-thieno[2, 3-c]pyran-3-carboxylic acid amide

Route 1 :
To a flask containing 2,2,6,6-tetramethyltetrahydro-4H-pyran-4-one (Int 2, 1 eq), cyanoacetamide (1 eq), sulfur (0.9 eq) and diethylamine (1.1 eq) are added. EtOH is then added and the resulting mixture is stirred at 40°C overnight. The reaction is diluted with water and partially concentrated by evaporation causing the precipitation of a solid that is separated by filtration. The cake is then washed with water and hexane to afford the desired product.
Alternative synthesis 1 of intermediate 3
Starting from 2,2,6,6-tetramethyltetrahydro-4H-pyran-4-one (Int 2, 1 eq), cyanoacetamide (1.1 eq) and morpholine (1.5 eq) are heated in EtOH at 80°C under inert atmosphere. After 6 h of heating, the mixture is cooled down, and sulfur (1.1 eq) is added. Next, the mixture is heated at 80°C overnight, then concentrated in vacuo and extracted with saturated NH4C1 and NaHCOs. The organic phase is subsequently dried over MgSO i, filtered and concentrated in vacuo. The residue obtained can finally be purified by column chromatography.
Alternative synthesis 2 of intermediate 3
A 20L glass reactor with a mechanical stirrer (400 rpm) and a reflux condenser is charged with 2,2,6,6-tetramethyltetrahydro-4H-pyran-4-one (Int 2) (1.466 kg, 9.01 mol, l eq) and 2-cyanoacetamide (1.363 kg, 1.8 eq.) followed by absolute EtOH (4.5 L) and morpholine (0.706 kg, 0.9 eq.). The resulting suspension is heated for 23 h at 75°C (internal temperature). After 23 h, sulfur (0.26 kg, 0.9 eq.) is added in one portion at 75°C and the resulting suspension is stirred further for 90 min after which the resulting solution is cooled to 20°C. Then, the entire solution is concentrated in vacuo (50 mbar / 45°C) to yield a solid residue. Water (13.5 L) is added in one portion at 75°C and the mixture is cooled to 22°C. Stirring (700 rpm at 22°C) is continued for 2.5h. The solids are separated by filtration, dried under vacuum suction, and subsequently in the vacuum oven at 40°C over 3d to obtain yield the desired product.
Intermediate 11: Dipyrazolo l,5-a;l ‘,5’-dJpyrazine-4,9-dione

[00213] 10 g (89 mmol) of pyrrazole carboxylic acid is suspended in toluene 100 mL at room temperature. Then, 2 equivalents of thionyl chloride are added, followed by a catalytic amount of DMF (0.5 ml). The mixture was stirred for lh at 75°C. After lh at 70 °C, the reaction was cooled to room temperature, the solid material was collected by filtration, washed with toluene and resuspended in DCM. Triethylamine (2 equivalents) was added and the suspension was stirred for 2h at room temperature. The product was collected by filtration, washed with DCM and dried at 40°C under vacuum to afford the desired product.
Example 4. Illustrative examples for the Preparation of the Compounds of Invention
Compound 2: N-(3-carbamoyl-5, 5, 7, 7 -tetramet yl-5 , 7-dihydro-4H-thieno[2, 3-c]pyran-2-yl)-lH-pyr zole-5-carboxamide

[00274] Intermediate 3 (15 g, 59 mmol) and 2H-pyrazole-3-carboxylic acid (9.9 g, 88 mmol) are suspended in DCM (250 mL). Mukaiyama reagent (2-chloro-l-methylpyridinium iodide) (18.1 g, 71 mmol), TEA (24.7 mL, 177 mmol) and DMAP (3.6 g, 29 mmol) are added. The reaction mixture is stirred at 40°C overnight and then cooled. The mixture is evaporated and the obtained crude is suspended in a 1 M HC1 solution. After stirring for 10 min, the suspension is filtered and obtained precipitate is isolated. This precipitate is re-suspended in a 0.1 M citric acid solution. Again, filtration gives a precipitate. A third trituration is done using ether as a solvent to give a precipitate after filtration. Finally, the precipitate (13.6 g) is suspended in EtOH (816 mL) and heated at reflux. To this suspension, 65 mL of DMF is added and a clear solution is obtained. The solution is concentrated to 275 mL and cooled at 0°C. A suspension is obtained, the solid is separated by filtration, and the cake is dried affording the desired product.
Alternative route

[00275] To a stirred (400 rpm) solution of 600 g (2.36 mol) of Intermediate 3 in DMAc (6 L), is added at ambient temperature 1.3 equivalents of Intermediate 11. To this resulting suspension, at room temperature, DIPEA (618 mL, 1.5 eq.) is added in small portions over a period of 5 min. The resulting suspension is heated to 80 °C and stirred for 18h at this temperature. The resulting mixture is cooled to 15°C and an aqueous saturated NH4C1 solution (7.5 L) is added over 30 minutes thus maintening the internal temperature between 15-24 °C. The resulting solid product is collected by filtration, and triturated with water (7.5 L) under mechanical stirring (600 rpm) for 30 min. The resulting suspension is filtered and the resulting solid is triturated in MTBE (8 L) under mechanical stirring for 45 minutes. The resulting solid is separated by filtration, and dried in a vacuum stove.
[00276] Finally, the solid is purified by hot trituration in ethanol. Therefore, the crude solid is suspended in absolute EtOH (16 L) for 1.5 h at 78 °C. The suspension is cooled to 20 °C and subsequently stirred for another hour. The solid product was collected by filtration, washed with 500 mL and again with 200 ml absolute EtOH, then dried to yield the desired product.
1H NMR PREDICT
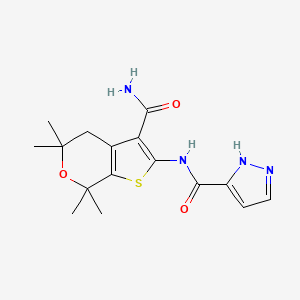
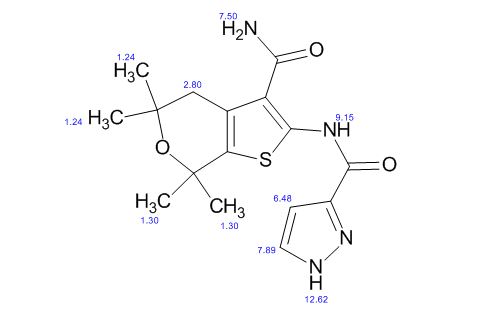
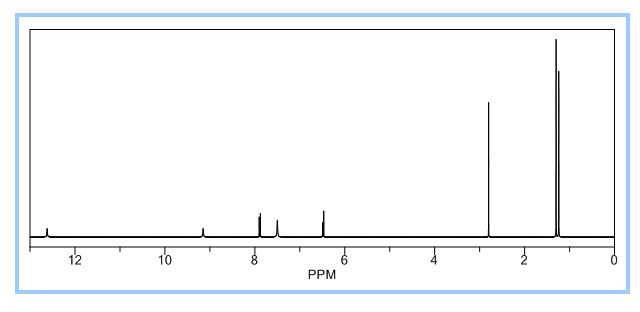
13 C NMR PREDICT
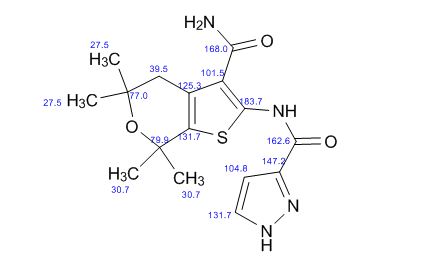
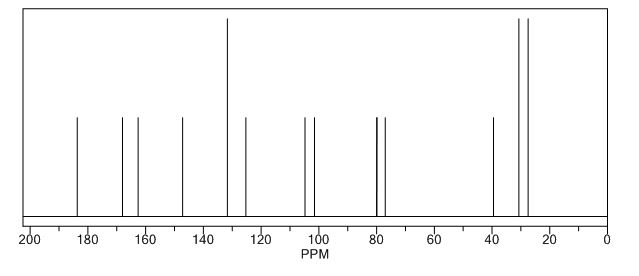
REFERENCES
First speaker at 1st disclosures is Steven Van der Plas of @GalapagosNV talking about a cystic fibrosis treatment #ACSSanFran
http://acsmeetings.cenmag.org/first-time-disclosures-of-clinical-candidates-at-acssanfran/?utm_source=Facebook&utm_medium=Social&utm_campaign=MeetingSF17
//////////////GLGP 1837
NC(=O)c2c3CC(C)(C)OC(C)(C)c3sc2NC(=O)c1ccnn1