















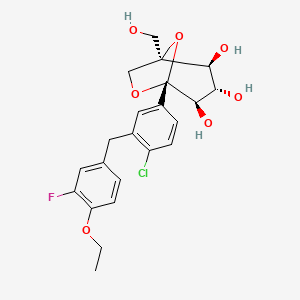

Henagliflozin, SHR-3824 ,
CAS 1623804-44-3
C22-H24-Cl-F-O7, 454.8756
PHASE 2 for the treatment of type 2 diabetes
China 20222, approvals 2022
HengRui (Originator)
| Jiangsu Hengrui Medicine Co Ltd |
UNII-21P2M98388; 21P2M98388; Henagliflozin; SHR3824; SHR-3824;

- HENAGLIFLOZIN PROLINE
- 4IO819SW6M
- 570.0 g/mol
- C27H33ClFNO9
- (1R,2S,3S,4R,5R)-5-[4-chloro-3-[(4-ethoxy-3-fluorophenyl)methyl]phenyl]-1-(hydroxymethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol;(2R)-pyrrolidine-2-carboxylic acid
In April 2016, Jiangsu Hengrui Medicine is developing henagliflozin (phase 2 clinical trial), a sodium-glucose cotransporter-2 (SGLT-2) inhibitor, for treating type 2 diabetes.
SGLT1 and SGLT2 inhibitors, useful for treating eg diabetes.
Henagliflozin proline is in phase II clinical trials by Jiangsu Hengrui (江苏恒瑞) for the treatment of type 2 diabetes.
1,6-dehydrated-1-C{4-chloro-3-[(3-fluoro-4-ethoxyphenyl)methyl]phenyl}-5-C-(hydroxymethyl)-β-L-idopyranose L-proline
(1 ^ 2345-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -1- (hydroxymethyl) 6,8 – alcohol dioxide
(1R,2S,3S,4R,5R)-5-[4-chloro-3-[(4-ethoxy-3-fluorophenyl)methyl]phenyl]-1-(hydroxymethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol
Henagliflozin is a pharmaceutical drug for the treatment of type 2 diabetes.[1] In China, it is approved for adult patients with type 2 diabetes to improve the glycemic control.[2][3]
Henagliflozin, like other drugs of the gliflozin class, inhibits the transporter protein sodium/glucose cotransporter 2 (SGLT2) which leads to a reduction in blood glucose levels.[4]
Shanghai Hengrui Pharmaceutical Co., Ltd., 上海恒瑞医药有限公司, Jiangsu Hengrui Medicine Co., Ltd., 江苏恒瑞医药股份有限公司, Less «
- 01 May 2015 Jiangsu HengRui Medicine Co. initiates enrolment in a phase I drug interaction trial in volunteers in China (NCT02500485)
- 12 Feb 2015 Jiangsu HengRui Medicine plans a phase I trial for Type-2 diabetes mellitus in China (NCT02366377)
- 01 Feb 2015 Jiangsu HengRui Medicine initiates enrolment in a phase I trial for Type-2 diabetes mellitus in China (NCT02366351)
Henagliflozin is a novel sodium-glucose transporter 2 inhibitor and presents a complementary therapy to metformin for patients with T2DM due to its insulin-independent mechanism of action. This study evaluated the potential pharmacokinetic drug-drug interaction between henagliflozin and metformin in healthy Chinese male subjects. 2. In open-label, single-center, single-arm, two-period, three-treatment self-control study, 12 subjects received 25 mg henagliflozin, 1000 mg metformin or the combination. Lack of PK interaction was defined as the ratio of geometric means and 90% confidence interval (CI) for combination: monotherapy being within the range of 0.80-1.25. 3. Co-administration of henagliflozin with metformin had no effect on henagliflozin area under the plasma concentration-time curve (AUC0-24) (GRM: 1.08; CI: 1.05, 1.10) and peak plasma concentration (Cmax) (GRM: 0.99; CI: 0.92, 1.07). Reciprocally, co-administration of metformin with henagliflozin had no clinically significant on metformin AUC0-24 (GRM: 1.09, CI: 1.02, 1.16) although there was an 11% increase in metformin Cmax (GRM 1.12; CI 1.02, 1.23). All monotherapies and combination therapy were well tolerated. 4. Henagliflozin can be co-administered with metformin without dose adjustment of either drug.
PATENT
PATENT
WO2012019496
https://www.google.com/patents/WO2012019496A1?cl=en
Example 4
(1 ^ 2345-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -1- (hydroxymethyl) 6,8 – alcohol dioxide


first step
1-ethoxy-2-fluoro – benzene
A mixture of 2-fluoro-phenol 4a (6.7 g, 60 mmol) was dissolved in 66 mL of acetone, was added iodoethane (6.3 mL,
78 mmol) and potassium carbonate (12.4 g, 90 mmol), at reflux in an oil bath for 5 hours. The reaction solution was concentrated under reduced pressure, was added 100 mL of ethyl acetate and 60 mL of water, separated, the aqueous phase was extracted with ethyl acetate (30 mLx2), the organic phases combined, dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure, to give the title product 1-ethoxy-2-fluoro – benzene 4b (6.9 g, red oil). yield: 82.1%.
MS m / z (ESI): 280.2 [2M + 1]
The second step
(5-bromo-2-chloro – phenyl) – (4-ethoxy-3-fluoro-phenyl) – methanone A mixture of 5-bromo-2-chloro – benzoyl chloride 2a (12.4 g, 48.8 mmol) was dissolved a 100 mL of dichloromethane was added 1-ethoxy-2-fluoro – benzene 4b (6.84 g, 48.8 mmol), cooled to 0 ° C, was added portionwise aluminum (5.86 g, 44 mmol) chloride, 16 h. Was added dropwise under ice-cooling to the reaction mixture 20 mL of 2 M HCl solution, separated, the aqueous phase was extracted with 30 mL of dichloromethane, and the combined organic phase was dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give the title The product (5-bromo-2-chloro – phenyl) – (4-ethoxy-3-fluoro-phenyl) – methanone 4c (12.7 g, yellow solid), yield: 72.6%.
MS m / z (ESI): 358.9 [M + l] Step
(5 – bromo-2-chloro – phenyl) – (4-ethoxy-3-fluoro-phenyl) – methanol (5-Bromo-2-chloro – phenyl) – (4-ethoxy -3 – fluoro – phenyl) -methanone 4c (12.7 g, 35.5 mmol) was dissolved in methanol and a 100 mL of tetrahydrofuran (ν: ν = 1: 1) mixed solvent, under an ice bath was added portionwise sodium borohydride (2.68 g, 70 mmol), and reacted at room temperature for 30 minutes. Add 15 mL of acetone, the reaction solution was concentrated under reduced pressure, 150 mL of ethyl acetate was added to dissolve the residue, washed with saturated sodium chloride solution (50 mLx2). The combined organic phase was dried over anhydrous magnesium sulfate, filtered, and concentrated under reduced pressure The filtrate, to give the title product (5-bromo-2-chloro – phenyl) – (4-ethoxy-3-fluoro-phenyl) – methanol 4d (12.7 g, orange oil), was used directly without isolation next reaction.
the fourth step
4 – [(5-bromo-2-chloro-phenyl) – methyl] Small-ethoxy-2-fluoro – benzene (5-bromo-2-chloro – phenyl) – (4-ethoxy -3 – fluoro – phenyl) methanol 4d (12.7 g, 35.3 mmol) was dissolved in a 100 mL of dichloromethane was added triethylsilane (16.9 mL, 106 mmol), was added dropwise boron trifluoride etherate (8.95 mL, 70.6 mmol ), for 3 hours. Was added 50 mL of saturated sodium bicarbonate solution, separated, the aqueous phase was extracted with ethyl acetate (100 mLx2), the organic phases combined, dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure, purified by silica gel column chromatography to elute B surfactant system resulting residue was purified to give the title product 4 – [(5-bromo-2-chloro – phenyl) methyl] -1-ethoxy-2-fluoro – benzene 4e (10 g, as a pale yellow oil ) yield: 82.4%.
1H NMR (400 MHz, CDC1 3 ): δ 7.33-7.27 (m, 3H), 6.95-6.90 (m, 3H), 4.14 (q, 2H), 4.01 (s, 2H), 1.49 (t, 3H)
the fifth step
(2 3R, 4S, 5 ^ 6R) -2- [4- chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6- (hydroxymethyl) – 2-methoxy – tetrahydro-pyran-3,4,5-triol
4 – [(5-bromo-2-chloro – phenyl) methyl] -1-ethoxy-2-fluoro – benzene 4e (7.36 g, 21.4 mmol) was dissolved in 30 mL of tetrahydrofuran, cooled to -78 ° C, was added dropwise a solution of n-butyllithium in hexane (10.27 mL, 25.7 mmol), at -78 ° C to react 1 hour, a solution of 20 mL (3R, 4S, 5R, 6R) -3,4,5 – tris (trimethylsilyloxy) -6- (trimethylsilyloxy) tetrahydropyran-2-one 2f (llg, 23.6 mmol) in tetrahydrofuran at -78 ° C under reaction 2 h, 2.8 mL of methanesulfonic acid and 71 mL of methanol, the reaction at room temperature for 16 hours. Was added 100 mL of saturated sodium carbonate solution, the reaction solution was concentrated under reduced pressure, to the residue was added 50 mL of saturated sodium chloride solution, extracted with ethyl acetate (100 mLx3), organic phases were combined, dried over anhydrous magnesium sulfate, filtered, The filtrate was concentrated under reduced pressure, purified by silica gel column chromatography with eluent systems resulting A residue was purified to give the title product (2 3R, 4S, 5 6R) -2- [4- chloro-3 – [(4-ethoxyphenyl 3-fluoro-phenyl) – methyl] phenyl] -6- (hydroxymethyl) -2-methoxy – tetrahydro-pyran-3,4,5-triol 4f (5.7 g, white solid ) yield: 58.3%.
1H NMR (400 MHz, CD 3 OD): δ 7.56 (s, 1H), 7.48 (dd, 1H), 7.37 (dd, 1H), 6.95-6.87 (m, 3H), 4.08-4.07 (m, 4H) , 3.91 (m, 1H), 3.93-3.73 (m, 2H), 3.56-3.53 (m, 1H), 3.45-3.43 (m, 1H), 3.30 (s, 2H), 3.08 (s, 3H), 1.35 (t, 3H)
The sixth step
(2 3R, 4S, 5 6R) -6- [(tert-butyl (dimethyl) silyl) oxymethyl] -2- [4-chloro-3 – [(4-ethoxy-3-fluoro – phenyl) methyl] phenyl] -2-methoxy – tetrahydro-pyran-3,4,5-triol the (2 3R, 4S, 5 6R) -2- [4- chloro-3- [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6- (hydroxymethyl) -2-methoxy – 4f tetrahydropyran-3,4,5-triol (5.7 g, 12.5 mmol) was dissolved in 50 mL of pyridine, followed by adding tert-butyldimethylsilyl chloride (2.26 g, 15 mmol) and 4-dimethylaminopyridine (305 mg, 2.5 mmol), for 16 hours. The reaction solution was concentrated under reduced pressure, was added 200 mL of ethyl acetate, washed with a saturated copper sulfate solution (50 mLx3). The combined organic phase was dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give the title product (2 3R, 4S, 5 6R) -6- [(tert-butyl (dimethyl) silyl) oxymethyl] -2- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -2-methoxy – tetrahydro-pyran-3,4,5-triol 4g (7.14 g, colorless oil), without isolation directly used for the next reaction.
Seventh Step
[[(2R, 3R, 4S, 5R, 6 ^ -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl yl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methoxy] – tert-butyl – dimethyl-silane (2 3R, 4S, 5 6R) -6- [(tert butyl (dimethyl) silyl) oxymethyl] -2- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -2-methoxy yl – tetrahydro-pyran-3,4,5-triol 4g (7.14 g, 12.5 mmol) was dissolved in 100 mL N, N- dimethylformamide was added 60% sodium hydride under ice-cooling (2.5 g , 62.5 mmol), and reacted at room temperature for 40 minutes completed the opening force, was added benzyl bromide (7.5 mL, 62.5 mmol), reaction of 16 hours. 20 mL of methanol, the reaction solution was concentrated under reduced pressure, was added 200 mL of ethyl acetate and 50 mL of water to dissolve the residue, separated, the aqueous phase was extracted with ethyl acetate (50 mL), the organic phase was washed with water (50 mL), washed with saturated sodium chloride solution (50 mL), the combined organic phase was dried over anhydrous magnesium sulfate , filtered, and the filtrate was concentrated under reduced pressure to give the title product [[(2R, 3R, 4S, 5R, 6 ^ -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4- ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methoxy] – tert-butyl – dimethylsilane 4h (10.5 g , yellow oil) yield: 99.8%.
Step Eight
[(2R, 3R, 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methanol
The [[(2R, 3R, 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl yl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methoxy] – tert-butyl – dimethylsilane 4h (10.52 g, 12.5 mmol) was dissolved in 50 mL of methanol dropwise add acetyl chloride CO.13 mL, 1.9 mmol), for 1 hour. The reaction solution was concentrated under reduced pressure, purified by silica gel column chromatography with eluent systems B resultant residue was purified to give the title product [(2R, 3R, 4S, 5R, 6 -3,4,5- tris-benzyloxy–6 – [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methanol 4i (7.6 g , yellow oil yield: 83.6%.
Step Nine
(2 ^ 3456 3,4,5-tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] – 6-methoxy – tetrahydropyran-2-carbaldehyde
Oxalyl chloride (1.17 mL, 13.6 mmol) was dissolved in 20 mL of dichloromethane, cooled to -78 ° C, were added dropwise 20 mL of dimethyl sulfoxide (1.56 mL, 21.9 mmol) in methylene chloride and 50 mL [(2R, 3R, 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methanol 4i (7.6 g, 10.45 mmol) in methylene chloride, and reacted at -78 ° C for 30 min, triethylamine (7.25 mL, 52.3 mmol), 2 hours at room temperature was added 50 mL 1 M HCl solution, separated, the organic phase was washed with saturated sodium chloride solution (50 mL x 2), the aqueous phase was extracted with dichloromethane (50 mL), the combined organic phase was dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give the title product (2 ^ 3456 3,4,5-tris-benzyloxy-6- [4-chloro-3 – [(4 – ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6-methoxy – tetrahydropyran-2-carbaldehyde 4j (7.58 g, colorless oil), was used directly without isolation next reaction.
The tenth step
(2S, 3 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl ] -2- (hydroxymethyl) -6-methoxy – tetrahydropyran-2-carbaldehyde
The (23456 3,4,5-tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] – 6-methoxy – tetrahydropyran-2-carbaldehyde 4j (7.6 g, 10.45 mmol) was dissolved in 80 mL 1,4- dioxane, followed by adding 15.8 mL 37% aqueous formaldehyde and sodium hydroxide solution (31.35 mL, 31.35 mmol), reacted at 70 ° C for 16 h. Add 50 mL of saturated sodium chloride solution, extracted with ethyl acetate (50 mLx4), the organic phase was washed with saturated sodium bicarbonate solution (50 mL), washed with saturated sodium chloride solution (50 mL), the combined organic phase was dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give the title product (23,456 benzyloxy-3,4,5-tris – 6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -2- (hydroxymethyl) -6-methoxy – tetrahydropyran – 2- formaldehyde 4k (7.9g, as a colorless oil), without isolation directly used for the next reaction.
Step Eleven
[(3 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] 2- (hydroxymethyl) -6-methoxy – tetrahydropyran-2-yl] methanol
The (23456 3,4,5-tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] – 2- (hydroxymethyl) -6-methoxy – tetrahydropyran-2-carbaldehyde 4k (7.9 g, 10.45 mmol) was dissolved in 50 mL of tetrahydrofuran and methanol (v: v = 2: 3) mixed solvent , was added sodium borohydride (794 mg, 20.9 mmol), for 30 minutes. Add a small amount of acetone, the reaction solution was concentrated under reduced pressure, purified by silica gel column chromatography with eluent systems resulting A residue was purified to give the title product, 5R, 6 -3,4,5-tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -2- (hydroxymethyl ) -6-methoxy – tetrahydropyran-2-yl] methanol 4m (l.ll g, colorless oil). yield: 14.1%.
Step Twelve
[(12345 ^ -2,3,4-tris-benzyloxy-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] 6,8-dioxa-bicyclo [3.2.1] octane-1-yl] methanol
The [(3S, 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] benzene yl] -2- (hydroxymethyl) -6-methoxy – tetrahydropyran-2-yl] methanol 4m (l.ll g, 1.46 mmol) was dissolved in 20 mL of dichloromethane, cooled to -10 ° C, was added trifluoroacetic acid (0.23 mL, 3 mmol), and reacted at room temperature for 2 hours. 20 mL of saturated sodium bicarbonate solution, separated, the aqueous phase was extracted with dichloromethane (20 mL> <2), and the combined organic phase was dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure, purified by silica gel column chromatography with eluent systems B resultant residue was purified to give the title product [(1 2 3 4R, 5 -2,3,4- tris-benzyloxy-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] 6,8-dioxa-bicyclo [3.2.1] octane-1-yl] methanol 4nC830 mg, colorless oil). yield: 78.3%.
MS m / z (ESI): 742.3 [M + 18]
Thirteenth Step
(12345-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -1- (hydroxymethyl) -6,8 dioxa-bicyclo [3.2.1] octane-2,3,4-triol
The [(1 2 3 4R, 5S) -2,3,4- tris-benzyloxy-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] benzene yl] -6,8-dioxa-bicyclo [3.2.1] octane-1-yl] methanol 4n (830 mg, 1.14 mmol) was dissolved in 20 mL of tetrahydrofuran and methanol (v: v = l: l) the a mixed solvent of o-dichlorobenzene was added (1.3 mL, 1 1.4 mmol) and Pd / C (500 mg, 10%), purged with hydrogen three times, the reaction for 3 hours. The reaction solution was filtered, rinsed with a small amount of ethyl acetate, the filtrate was concentrated under reduced pressure, purified by silica gel column chromatography with eluent systems resulting A residue was purified to give the title product (1S, 2 3S, 4R, 5 -5- [ 4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -1- (hydroxymethyl) -6,8-dioxa-bicyclo [3.2.1] octane-2,3,4-triol 4 (420 mg, white solid), yield: 81.0% MS m / z (ESI):. 472.2 [m + 18]
1H NMR (400 MHz, CD 3 OD): δ 7.47 (s, 1H), 7.42-7.35 (m, 2H), 6.95-6.87 (m, 3H), 4.16-4.14 (m, 1H), 4.06-4.02 ( m, 4H), 3.85-3.70 (m, 2H), 3.67-3.54 (m, 4H), 1.37 (t, 3H)
References
- Weng J, Zeng L, Zhang Y, Qu S, Wang X, Li P, et al. (August 2021). “Henagliflozin as add-on therapy to metformin in patients with type 2 diabetes inadequately controlled with metformin: A multicentre, randomized, double-blind, placebo-controlled, phase 3 trial”. Diabetes, Obesity & Metabolism. 23 (8): 1754–1764. doi:10.1111/dom.14389. PMID 33769656.
- Wang G (17 February 2022). “Monthly Report: New Drug Approvals in China, January 2022”. BaiPharm.
Henagliflozin Proline Tablets
- “Henagliflozin – Jiangsu HengRui Medicine”. AdisInsight. Springer Nature Switzerland AG.
- He X, Liu G, Chen X, Wang Y, Liu R, Wang C, et al. (July 2023). “Pharmacokinetic and Pharmacodynamic Interactions Between Henagliflozin, a Novel Selective SGLT-2 Inhibitor, and Warfarin in Healthy Chinese Subjects”. Clinical Therapeutics. 45 (7): 655–661. doi:10.1016/j.clinthera.2023.06.002. PMID 37451912.
 |
|
| Clinical data | |
|---|---|
| Trade names | Rui Qin; 瑞沁 |
| Other names | SHR3824; SHR-3824 |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| UNII | |
| Chemical and physical data | |
| Formula | C22H24ClFO7 |
| Molar mass | |
////////Henagliflozin, SHR-3824 , PHASE 2, type 2 diabetes, UNII-21P2M98388, 21P2M98388, SHR 3824, SHR3824, approvals 2022, china 2022, Henagliflozin proline
CCOc1ccc(cc1F)Cc2cc(ccc2Cl)[C@]34[C@@H]([C@H]([C@@H]([C@](O3)(CO4)CO)O)O)O
SYN
Synthesis 2024, 56, 906–943
Henagliflozin (12) (also known as SHR3824), developed by Lexicon Pharmaceuticals (Princeton, NJ, USA), is a potent and selective SGLT inhibitor administered orally. In 2013, the first synthetic route for the preparation of henagliflozin (12) was described and claimed by two pharmaceutical companies: Shanghai Hengrui Pharmaceutical Co., Ltd., and Jiangsu Hengrui Medicine Co., Ltd. Several other C-aryl-glucoside-type derivatives were prepared and registered in the United States under patent application number US8609622B2.67 Among these derivatives, the synthesis of henagliflozin (12) was carried out using a thirteen-step process, resulting in an overall yield of 3% (Schemes 40 and 41). The process consisted of the formation of the key intermediate 215 starting from commercially available 2-fluorophenol (211). In the first step, phenolic compound 211 was converted into 212 in 82% yield using ethyl bromide and po
tassium carbonate in acetone. The Friedel–Crafts reaction of acid chloride 26c′ using AlCl3 in DCM afforded intermediate 213 in 72% yield, which was further reduced to 214 using NaBH4 in a mixture of THF/MeOH. Without further isolation, the reduction of 214 was carried out using Et3SiH and BF3·Et2O in DCM to give 215 (Scheme 40). The intermediate 215 was taken forward for lithium halogen exchange using n-BuLi followed by addition of the lithiated compound to O-silyl-protected compound 22 at
low temperature to afford a lactol intermediate. The obtained lactol intermediate was protected using
MsOH/MeOH to give the desired product 216 in 58% yield. Under the above conditions, deprotection of the O-silylgroups of the C-glucoside 22 was also observed. Further, under basic conditions, the secondary hydroxy group of 216 was silyl protected using tert-butyldimethylsilyl chloride (TBSCl) and DMAP to afford compound 217, which was treated with NaH and BnBr to give benzylated compound
218 in excellent yield. In methanol solution, deprotection of the silyl protecting group of compound 218 using acetylchloride afforded 219. Swern oxidation of the hydroxy compound 219 in the presence of oxalyl chloride and DMSO gave intermediate 220, which was used for the next step without isolation. The crude compound 220 was treated with NaOH and 37% formaldehyde solution to afford 221.
Dihydroxy intermediate 222 was then obtained in low yield via reduction of the aldehyde group of compound 221 with sodium borohydride in THF/MeOH mixture. Next, treatment of 222 with trifluoroacetic acid gave compound 223. Debenzylation of compound 223 was carried out by Pd/C
catalytic hydrogenation to afford the final product henaglifozin (12) (Scheme 41).
The highlight of the synthesis is the design of the route with minimal isolation stages and intermediates possessing unstable functional groups were subjected to subsequent transformations in situ. The drawbacks of the above synthetic process are the use of a protection and deprotection
strategy that led to low throughput and the final compound being obtained in low yield. Reduction of the aldehyde in 221 mediated by sodium borohydride resulted in a poor yield of product 222, and this procedure is not recommend ed for scale-up due to safety concerns. Additionally, the use
of palladium in the last step of the synthesis involves the risk of this toxic metal leaching into the final product. To address the issue with the discovery route, Yongjun and co-workers reported an alternative approach to obtain compound 12 (Scheme 42).68 The authors published the synthesis of henagliflozin proline (12a) starting from TMS protected D-glucolactone 22 and aglycone intermediate The diol 226 was obtained after carrying out a disproportionation reaction on the aldehyde using paraformaldehyde under strong alkaline conditions. Intramolecular etherification of diol 226 using 30% HCl gave henagliflozin
(12) in 95% yield, which was further treated with L-proline to give henagliflozin proline monohydrate 12a. The authors reported several advantages such as easy steps, cost-effective procedures, simple product purification and an overall method that was amenable for commercialization. This Addition of the aglycone intermediate 215 was carried out with 22 followed by mesylation of the OH group to provide 216 in 65% yield. Further, all the secondary hydroxy groups of intermediate 216 were selectively protected us ing TMSCl, imidazole and PPTS to give 224 in 95% yield. The free primary hydroxy group of 224 was oxidized using pyridine sulfur trioxide in triethylamine and DMSO to afford process involves 10 steps and gave an overall yield of 22% of henagliflozin proline (12a) (Schemes 40 and 42)
REF 67, 68
(67) Yang, F.; Tang, P. C.; Dong, Q.; Tu, W.; Fan, J.; Guan, D.; Shen, G.;Wang, Y.; Yuan, J.; Zhang, L. US8609622B2, 2013.
(68) Chun, K.; Peng, Z.; Qichao, L.; Bo, Z.; Zhen, W.; Guorong, Z.;Yongjun, T. CN 112375087A, 2020.


.