
















LEVALBUTEROL TARTRATE
Levosalbutamol
cas 661464-94-4
4-[(1R)-2-(tert-butylamino)-1-hydroxyethyl]-2-(hydroxymethyl)phenol;(2R,3R)-2,3-dihydroxybutanedioic acid
MW 628.7, C30H48N2O12
- Xopenex HFA
- Levosalbutamol tartrate
- ADS4I3E22M
- UNII-ADS4I3E22M
- Levosalbutamol tartrate(levalbuterol) is the R-enantiomer of the short-acting β2-adrenergic receptor agonist salbutamol.
Levalbuterol Tartrate is the tartrate salt form of levalbuterol, the R-enantiomer of the short-acting beta-2 adrenergic receptor agonist albuterol, with bronchodilator activity. Levalbuterol selectively binds to beta-2 adrenergic receptors in bronchial smooth muscle, thereby activating intracellular adenyl cyclase, an enzyme that catalyzes the conversion of adenosine triphosphate (ATP) to cyclic-3′,5′-adenosine monophosphate (cAMP). Increased cAMP levels cause relaxation of bronchial smooth muscle, relieve bronchospasms, improve mucociliary clearance and inhibit the release of mediators of immediate hypersensitivity from cells, especially from mast cells.
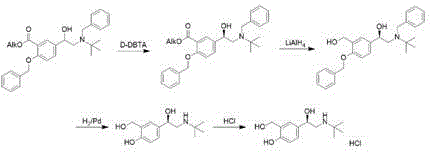
British patent document GB1298494A firstly discloses synthesis of levosalbutamol, which comprises the steps of carrying out crystallization resolution by using D- (+) -dibenzoyl tartaric acid, carrying out ester reduction reaction, and removing two benzyl protecting groups to obtain levosalbutamol, wherein the process route is as follows:
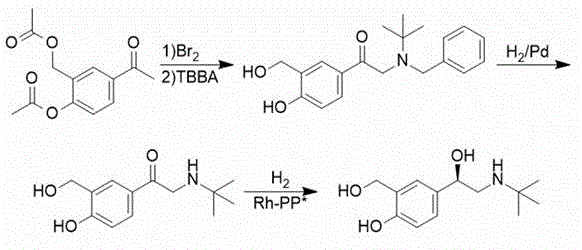
chinese patent CN1705634A, and using rhodium and chiral bidentate phosphine ligand combination, levosalbutamol can be obtained with good yield and good optical purity on a technical scale. The disadvantages are that the toxicity of the reagent is high, the hydrogenation risk is high, and the process route is as follows:
SCHEME

PATENTS
MX2012014342
IN2009MU01097
IN2007CH01847
US20040115136
CN1382685
PATENT
https://patents.google.com/patent/CN114539077A/en
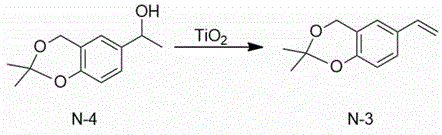
The technical scheme adopted by the invention is as follows: 1) 1- (2, 2-dimethyl-4H-benzo [ d ] [1,3] dioxin-6-yl) ethanol and titanium dioxide are used as initial raw materials, a solvent-free system is adopted, and 2, 2-dimethyl-6-vinyl-4H-benzo [ d ] [1,3] dioxin is synthesized through dehydration.
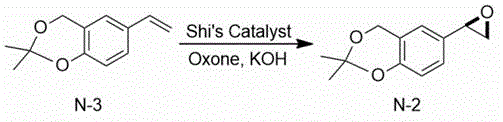
Then, the 2, 2-dimethyl-6-vinyl-4H-benzo [ D ] [1,3] dioxin is subjected to epoxidation under the combined action of 1,2:4, 5-di-O-isopropylidene-BETA-D-erythro-2, 3-dione-2, 6-pyranose (Shi’s Catalyst), Oxone and potassium hydroxide to obtain (R) -2, 2-dimethyl-6- (oxirane-2-yl) -4H-benzo [ D ] [1,3] dioxin.
Reacting and condensing (R) -2, 2-dimethyl-6- (epoxy ethane-2-group) -4H-benzo [ D ] [1,3] dioxin and tert-butylamine in ethanol, and salifying with D- (+) -malic acid to obtain (R) -2- (tert-butylamine) -1- (2, 2-dimethyl-4H-benzo [ D ] [1,3] dioxin-6-group) ethanol D- (+) -malate.
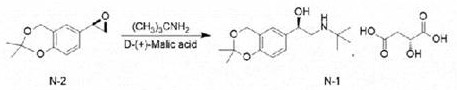
And (R) -2- (tert-butylamine) -1- (2, 2-dimethyl-4H-benzo [ D ] [1,3] dioxin-6-yl) ethanol D- (+) -malate is subjected to hydroaminolysis and reacts with hydrogen chloride ethanol to prepare the levosalbutamol hydrochloride.
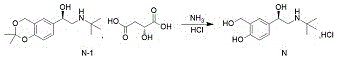
The invention discloses a novel method for synthesizing levalbuterol hydrochloride, wherein the synthesis of a key intermediate is novel without cyclization oxidation, the total yield is 85-90%, and the method is higher than that of the conventional method. The process is convenient to operate, the raw materials are economical, and the method is suitable for large-scale industrial production.
EXAMPLE 12 preparation of 2, 2-dimethyl-6-vinyl-4H-benzo [ d ] [1,3] dioxine
A1000 mL flask was charged with 208g (1.0 mol) of 1- (2, 2-dimethyl-4H-benzo [ d ] [1,3] dioxin-6-yl) ethanol, which was accurately weighed, and stirring was started. Then slowly adding 16g (0.2 mol) of titanium dioxide, installing a water separator and a water flow pipe, starting heating until the internal temperature is kept at 120-130 ℃, and stirring for 12 hours. After the reaction is finished, the temperature is reduced to below 50 ℃, the water separator is removed, the reduced pressure distillation device is changed, and 120 ℃ (less than 100 Pa) fraction is collected to obtain 180.7g of 2, 2-dimethyl-6-vinyl-4H-benzo [ d ] [1,3] dioxin with the yield of 95%.
Mass spectrum: EI (m/z): 190; hydrogen nuclear magnetic resonance spectroscopy:1HNMR(400MHz,CDCl3)δ7.55(d,J=4Hz,1H),7.11(s,1H),6.87(d,J=4Hz,1H),6.65~6.60(m,1H),5.63~5.60(m,1H),5.19~5.5(m,1H),4.59(s,2H),1.49(s,6H)。
EXAMPLE 2 Synthesis of (R) -2, 2-dimethyl-6- (oxiran-2-yl) -4H-benzo [ d ] [1,3] dioxine
A clean 5000mL three-neck flask is taken, 180.5g (0.95 mol) of the compound
2, 2-dimethyl-6-vinyl-4H-benzo [ D ] [1,3] dioxin obtained in the example 1 is added, 2000mL of acetonitrile is added for dissolution, 1,2:4, 5-di-O-isopropylidene-BETA-D-erythro-2, 3-dione-2, 6-pyranose 49.1g (0.19 mol) is added, potassium monopersulfate (Oxone) 876g (1.43 mol) is added under stirring, a proper amount of potassium hydroxide is added after the addition is finished, the pH of the system is kept between 10 and 11, and the stirring reaction is continued at 25 ℃ for 8 to 12 hours. After the reaction, the mixture was slowly poured into 2000ml of purified water prepared in advance, stirred sufficiently for 30min, and then was allowed to stand for layering, and the organic layer was collected. 2000ml of dichloromethane is added for extraction, organic layers are combined and washed by saturated sodium chloride solution, the organic layer is dried by adding anhydrous sodium sulfate and concentrated to dryness to obtain 196g of crude colorless liquid with the yield of 100 percent.
Mass spectrum: EI (m/z): 207; hydrogen nuclear magnetic resonance spectroscopy:1HNMR(400MHz,CDCl3)δ7.25(s,1H),7.18(d,J=4Hz,1H),6.85(d,J=4Hz,1H),4.59(s,2H),3.85~3.81(m,1H),2.96~2.71(m,2H),1.49(s,6H)。
EXAMPLE 3 preparation of (R) -2- (tert-butylamine) -1- (2, 2-dimethyl-4H-benzo [ D ] [1,3] dioxin-6-yl) ethanol D- (+) -malate salt
A clean 5000mL three-neck flask is taken, the compound (R) -2, 2-dimethyl-6- (oxiranyl-2-yl) -4H-benzo [ d ] [1,3] dioxin obtained in the example 2 is added, 196g (0.95 mol) of the clean 5000mL three-neck flask is taken, 1000mL of ethanol is added for dissolution, 80.4g (1.1 mol) of tert-butylamine is added, stirring is started, heating is carried out till reflux, reaction is carried out for 3H, and the progress of the reaction is detected by TLC. After the reaction is finished, 127g (0.95 mol) of D- (+) -malic acid is added in batches, and stirring and refluxing are continued for 2h after the addition is finished. And then cooling to 5-15 ℃, precipitating a large amount of solid, stirring for 3H, filtering, washing the filter cake with ethanol, collecting the filter cake, and drying to obtain (R) -2- (tert-butylamine) -1- (2, 2-dimethyl-4H-benzo [ D ] [1,3] dioxin-6-yl) ethanol D- (+) -malate 372g of white solid with the yield of 94.7%.
Mass spectrum: ESI (m/z): 280.1, respectively; hydrogen nuclear magnetic resonance spectroscopy:1HNMR(400MHz,d-DMSO)δ7.25(s,1H),7.18(d,J=4Hz,1H),6.85(d,J=4Hz,1H),4.90~4.76(m,2H),4.59(s,2H),4.44~4.40(m,2H),3.65(br,2H),3.15~2.90(m,2H),2.77~2.52(m,2H),2.03(s,1H),1.50(s,6H),1.27(s,9H)。
EXAMPLE 4 preparation of L-salbutamol hydrochloride
A5000 mL beaker was charged with 372g of the compound (R) -2- (tert-butylamine) -1- (2, 2-dimethyl-4H-benzo [ D ] [1,3] dioxin-6-yl) ethanol D- (+) -malate salt obtained in example 3, 1500mL of purified water was added, and the mixture was stirred to dissolve it, followed by addition of 1500mL of dichloromethane and cooling in an ice bath. Slowly adding a proper amount of concentrated ammonia water under stirring to adjust the pH value of the water phase to 9-10, continuously stirring for 30min, and standing for layering. Separating and collecting organic layer, adding 1000ml of dichloromethane into water layer, stirring for 10min, standing and demixing. Separating and collecting organic layers, combining the organic layers, adding 2000ml of saturated sodium chloride solution into the organic layers, stirring for 30min, standing for layering, collecting the organic layers, adding a proper amount of anhydrous sodium sulfate, drying, filtering, washing with dichloromethane, and collecting filtrate.
And (3) carrying out rotary evaporation and concentration on the filtrate to about 1500mL, transferring the concentrated filtrate into a 5000mL three-neck bottle, and placing the three-neck bottle in an ice bath to cool the three-neck bottle to 5-15 ℃. About 110g of 30% hydrogen chloride ethanol solution is dropwise added under stirring, and after the dropwise addition is finished, 2000mL of methyl tertiary butyl ether is dropwise added under stirring, so that a large amount of white solid is precipitated. And after the addition is finished, continuously stirring for 3 hours at the temperature of 5-15 ℃, filtering, adding methyl tert-butyl ether into a filter cake for washing, collecting the filter cake, and drying to obtain 241.5g with the yield of 97.3%. Through HPLC analysis, the purity is 99.95%, and the isomer content is not detected, as shown in figures 1-4. The total yield of the four-step reaction is 87.5 percent.
Publication numberPriority datePublication dateAssigneeTitle
CN1413976A *2002-09-132003-04-30苏州君宁新药开发中心有限公司New process for preparing levo-albuterol
US20050261368A1 *2004-05-202005-11-24Valeriano MerliPreparation of levalbuterol hydrochloride
CN103951568A *2014-05-192014-07-30苏州弘森药业有限公司New process for synthesizing salbutamol and sulfate of salbutamol
CN104557572A *2014-12-302015-04-29上海默学医药科技有限公司Levalbuterol intermediate and levalbuterol hydrochloride synthesis method
CN110963929A *2019-11-262020-04-07安徽恒星制药有限公司Preparation method of salbutamol hydrochloride suitable for industrial production
CN113227113A *2018-12-202021-08-06帝斯曼知识产权资产管理有限公司Improved synthesis of epoxidation catalysts
CN113801029A *2020-06-162021-12-17盈科瑞(天津)创新医药研究有限公司Preparation method of levalbuterol hydrochloride
//////////Levosalbutamol, LEVALBUTEROL TARTRATE, Xopenex HFA, Levosalbutamol tartrate, ADS4I3E22M, UNII-ADS4I3E22M